
Development of novel therapies for Huntington’s disease: hope and challenge1
Introduction
Huntington's disease (HD) is a devastating neurological disorder without effective treatment. It is characterized by involuntary movement, cognition impairment, and psychological disturbance. The disease affects 5-10 out of 10 000 people in western countries. HD is a fatal neurological disorder and death usually occurs approximately 20 years after the onset of symptoms. Therefore, there is an urgent need for developing effective therapies for HD. The genetic cause of HD was uncovered in 1993[1]. The disease is linked to an expansion of polyglutamine in the novel protein, huntingtin. Since 1993, extensive research efforts have been made to understand pathogenic mechanisms of mutant huntingtin. Multiple molecular mechanisms seem to be involved in HD pathogenesis[2]. Researchers are actively investigating possible effective therapies based on our current understanding of the molecular pathology of HD. One approach that has been explored is high-throughput screening for small molecules affecting specific molecular targets. Potential targets for high-throughput chemical screenings include molecular chaperones, caspases, the ubiquitin/proteasome proteases, transcription factors, and the offending polyglutamine-containing proteins themselves. Much attention has been focused on screening for drugs that prevent aggregation of huntingtin with expanded polyglutamine tract. In vivo screenings are also being planned or are under-way using transgenic HD mice and Drosophila[3]. The other approach is to use recent advances in molecular biology of neurotrophic factors, neuronal tissue transplantation and cell engineering in HD therapeutic developments with the aim of retarding or reversing HD pathology, but without complete understanding of the molecular pathogenic mechanisms. Both approaches are moving at a fast pace and we expect new therapies will be available clinically soon. This paper reviews current treatments for clinical symptoms of HD and the development of novel therapies targeted at the molecular pathogenic mechanisms of mutant huntingtin.
Developing novel therapies aimed at mutant huntingtin
The expansion of the polyglutamine tract in huntingtin causes the "gain-of-function"? mutation. Gained functions include misfolding and aggregation, abnormal protein interactions, and dysregulation of transcription conferred on mutant huntingtin. The ideal therapy should correct these acquired properties in mutant huntingtin.
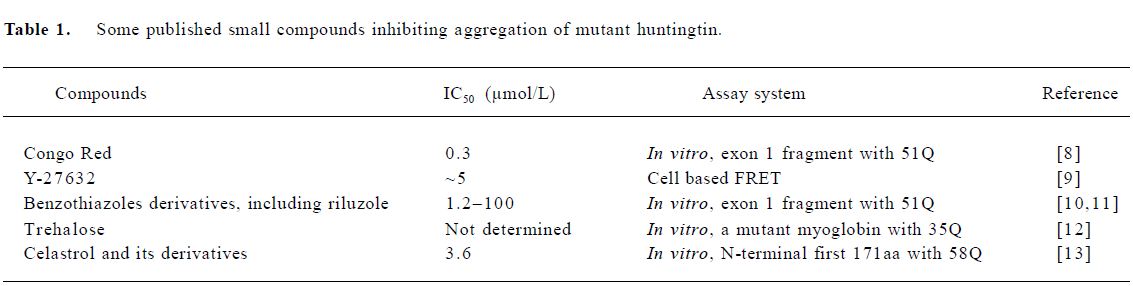
Compounds inhibit mutant huntingtin aggregation The in vitro aggregation of huntingtin involves a conformational change of the polyglutamine segment from a random coil to an amyloid structure, and is in some ways paralleled in cell culture by the formation of cytoplasmic aggregates and nuclear inclusions[4-6]. Neuronal inclusions containing amino-terminal fragments have also been detected in HD brains and have become a hallmark of HD pathology. The role of huntingtin aggregates and inclusions in pathogenesis is still being debated, as they may or may not correlate to cell death[7]. Whether the formation of huntingtin aggregates in the brain is the cause or merely the consequence of the disease is unclear. However, aggregation of mutant huntingtin has been shown to be related to HD progress. Keeping mutant huntingtin from aggregation would provide a way to prevent the progression of the disease.
Several laboratories have screened the NIH (National Institutes of Health) Small Compounds Library with in vitro aggregation assay using purified bacterial-expressed N-terminal huntingtin fragments or in vivo cell-based aggregation assay. Table 1 shows some published small compounds that directly inhibit aggregation of mutant huntingtin.
These direct aggregation inhibitors have been tested in various HD models such as cell culture, HD Drosophila and transgenic HD mice. Congo Red showed protective effects on survival, weight loss, and motor function even after the onset of symptoms of HD in R6/2 transgenic mice. The underlying mechanisms of Congo red include keeping mutant huntingtin fragments from aggregating and increasing the accessibility of huntingtin by proteasome[14]. Y-27632, an inhibitor of the Rho-associated kinase p160ROCK, prevents polyglutamine protein aggregation in cultured cells and reduces neurodegeneration in an HD Drosophila model[9]. Among benzothiazoles, the strongest inhibitors of mutant huntingtin aggregation, with IC50 ranging from 1.2-11.0 μmol/L in the in vitro assay, are very toxic to the cells. But two weak inhibitors with lower IC50 are nontoxic and effective in reducing the aggregation of mutant huntingtin fragments in a cell-based assay. Riluzole, a glutamate release inhibitor, showed neuroprotective effects in R6/2 transgenic HD mice by prolonging their survival and delaying progressive weight loss, but had no effect on motor coordination, locomotor activity, and the formation of neuronal intranuclear inclusions (NII) [11]. Trehalose, a disaccharide, reduced aggregation of mutant huntingtin fragments in cells and NII in R6/2 HD transgenic mice. It was proposed that trehalose binds to expanded polyglutamines to prevent aggregation. R6/2 HD mice treated with trehalose showed improved motor function and extended lifespan[12]. Celastrol, a natural bioactive compound, was effective in reversing the abnormal cellular localization of full-length mutant huntingtin observed in striatal cells derived from HdhQ111/Q111knock in mice[13].
Compounds identified by cell-free aggregation assay are usually either non-effective in the cell-based aggregation assay or toxic to cells. For example, many active benzo-thiazoles[10], compounds identified by cell-based assay usually reduce aggregation of mutant huntingtin fragments by indirect mechanisms, such as Y-27632[9]. Combining both cell-free and cell-based assays would make a more reliable prediction of potential small molecules for further drug development. Most experiments used for screening and testing compounds, including in vitro aggregation assays, cell-based aggregation assays, HD Drosophila and HD transgenic mice, are based on small N-terminal huntingtin fragments with an expanded ployglutamine tract. Still uncertain is the effectiveness of these compounds in transgenic HD mice, particularly when full-length mutant huntingtin is expressed. As more and more laboratories are conducting screening and developing aggregation inhibitors, it is expected that more and more effective compounds will be identified.
In addition to direct aggregation inhibitors, inhibitors of cell toxicity caused by over-expression of N-terminal fragments with expanded ployglutamine tract were also screened from bioactive compounds[15]. Caspase inhibitors and cannabinoids showed protective effects with EC50 of 10 μmol/L and 30 μmol/L, respectively. Induction of expression of heat shock proteins with geldanamycin also inhibited huntingtin aggregation and huntingtin induced cellular toxicity[16].
Transglutaminase inhibitor It has been shown in vitro that transglutaminase (TGase) can use huntingtin as a substrate to cross-link huntingtin molecules[17], and gives preference to huntingtin with polyglutamine more than 36 repeat[18]. TGase activity was found to have increased in HD postmortem brains, and aggregated huntingtin catalyzed by TGase were found in NII[18]. TGase provides an additional mechanism for the formation of aggregation of mutant huntingtin. This suggests that TGase might play a role in HD pathogenesis and, therefore, is a potential therapeutic target. Cystamine is an inhibtor of TGase[19]. Treatment of R6/2 transgenic HD mice with cystamine showed a small but significant neuroprotective effect with improvement of motor function, survival and loss of bodyweight. However, whether it can reduce mutant huntingtin aggregates is not clear as contradictory results were reported[20]. The underlying mecha-nism(s) is complicated as cystamine is also an antioxidant and increases transcription of certain neuroprotective chaper-ones, HDJ1/Hsp40[21].
Protease inhibitors Recent findings showed that huntingtin can be cleaved by proteases, including cas-pases[22,23], calpain[24,25], and aspartyl protease[26]. N-terminal huntingtin fragments were found in postmortem HD patients? brains[27], and YAC transgenic mice that expressed full-length human huntingtin with expanded polyglutamine tract[28]. Because caspase and calpain-mediated partial cleavage of mutant huntingtin promotes huntingtin aggregation and cellular toxicity, inhibitors of huntingtin partial cleavage might have therapeutic values.
The potential capases cleavage sites are located within amino acids 513?586 and only capases 2, 3, and 6 were found to be effective[23]. Calpain cleaves huntingtin at amino acids 469 and 536[25]. Aspartic endopeptidases release two distinct mHtt fragments, cp-A and cp-B, by cleaving huntingtin between amino acids 104-114. Cp-A is the major fragment that aggregates in the nucleus[26]. In the cell based assays, caspase-resistant and calpain-resistant htts with expanded polyglutamine tract were reported to decrease aggregate formation and reduce cytotoxicity[23,25]. Caspase inhibitors, z-VAD-fmk and z-DEVD-fmk, can prevent cleavage of hun-tingtin by caspases and reduce cytotoxicity caused by expanded polyglutamine tract[23,29]. As N-terminal huntingtin fragments produced by caspase and calpain mediated cleavage are localized in the nucleus and prone to aggregate, this might contribute to HD pathogenesis. Protease inhibitors could reduce N-htt fragments and, in turn, prevent or delay disease progression. A few studies demonstrate that the caspase inhibitor minocycline was able to inhibit huntingtin aggregation, retard disease progress and prolong the lifespan of HD mice[30,31]. So far, there is no good inhibitor for calpain and aspartic endopeptidases that have been developed and tested against mutant huntingtin.
Apoptosis or programmed cell death is implicated in HD pathogenesis by the observations of the elevation of cytochrome c in the cytoplasm[32] and activation of caspases 1, 3, 8, and 9[32-34]. Thus, the apoptosis pathway is a reasonable target for HD therapy. In one study, intracerebroventricular administration of z-VAD-fmk, as an inhibitor of apoptosis, for 4 weeks starting at the age of 7 weeks improved survival of R6/2 mice by 24.8%[34]. However, in another study, administration of other apoptosis inhibitors, YVAD-fmk and DEVD-fmk individually, showed no effects at all, though they can extend the lifespan of R6/2 mice by 17.2%[30,31]. As therapeutic agents, caspase inhibitors might act on two targets: mutant huntingtin to prevent the release of toxic N-terminal fragments and apoptosis to prevent neurons from death caused by mutant huntingtin.
Histon deacetylase (HDAC) inhibitors Huntingtin interacts with several important transcription factors, including cAMP-response element binding protein (CBP), TATA box binding protein (TBP), specificity protein-1 (SP1), nuclear factor-κB (NF-κB) [35-37]. Some of these are reportedly found in huntingtin aggregates. Altered gene expression was found in the R6/2 transgenic HD mice at the ages of 6 and 12 months[38], and in full-length huntingtin transgenic mice[39]. Abnormal transcription might be caused by direct interactions between mutant full-length huntingtin or its fragments and transcription factors, which in turn change the normal transcription mediated by these transcription factors. Mutant huntingtin can affect transcription by altered binding to transcription factors not recruiting them into aggregates. This means that altered transcription is an early event in HD pathogenesis caused by soluble mutant huntingtin, either full-length or fragmented before forming aggregates. This notion is supported by several lines of evidence: (1) no CBP, TBP, or SP1 could be found in huntingtin inclusions in several HD mouse models and they maintained normal cellular levels in these mice[40]; (2) less SP1 was found to bind to DNA in the presence of mutant huntingtin in vitro[36] and in vivo[41].
Reversal of the altered transcription might be a way to retard HD progression. Inhibitors of histone deacetylase (HDAC) can increase gene transcription and have been examined as a potential therapy in both HD Drosophila and transgenic R6/2 HD mice. Suberoylanilide hydroxamic acid (SAHA), a selective HDAC inhibitor, reduced neurodegener-ation in HD Drosophila[42]. In R6/2 mice, SAHA increased histone acetylation and ameliorated motor deficits. However, it did not affect the aggregation and loss of brain weight[43]. Another HDAC inhibitor, sodium butyrate, improved survival of R6/2 mice in a dose-dependent manner by up to 21.7% when administered intraperitoneally at a dose of 200-10 000 mg•kg-1•d-1. This treatment increased acetylation of histon 3, histon 4, and SP1, improved motor performance and bodyweight loss, and decreased atrophy of the brain and striatum. But sodium butyrate had no effect on the formation of immunoreactive huntingtin aggregates[44].
Gene therapy Gene therapy is another potential therapeutic method for HD in addition to the small compounds described above. Intracellular antibodies (intrabodies) and RNA interference (RNAi) are 2 potential methods that could be used for gene therapy of HD. Intrabodies are a powerful tool to study protein functions in vivo and also a potential therapeutic approach delivered by gene therapy[45]. This is achieved by expressing single-chain variable region fragments (svFv or sFv) of the antibody intracellularly to bind an intracellular target[46]. One intrabody selected from a human sFv phage display library, which is specific for the first 17 amino acids of human huntingtin, dramatically reduced the aggregates formation when cotransfected with huntingtin, with expanded polyglutamine tract and a GFP tag[47]. A recent study reported that an intrabody consisting of only a single light chain variable domain selected from a non-immune human antibody library against the first 20 N-terminal amino acids of human huntingtin and affinity matured with yeast surface display method, inhibited mutant huntingtin aggregation in both cell-free and cell-based assays[48]. The small size of this single-domain intrabody enables it to diffuse into the nucleus readily. Given its small size and human origin, it should be a more practical approach in gene therapy. The recognition site of the intrabody determines whether the intrabody has beneficial effects. In addition to intrabodies recognizing the first 17 or 20 amino acids of huntingtin, the intrabody recognizes the polyproline domains of huntingtin inhibited aggregation and reduces the cell death caused by mutant huntingtin. However, an intrabody recognizing polyglutamine region actually promoted aggregation and apoptosis[49].
Recently developed RNAi technology provides a power-ful way to reduce target gene expression in cell cultures and in brains[50]. To explore its potential therapeutic effects in polyglutamine diseases, double-stranded RNA (dsRNA), which induces RNA interference, was used to test against polyglutamine induced cell toxicity[51]. dsRNA can cause sequence-specific, not allele-specific inhibition of huntingtin translation. In mammalian cells, dsRNA reduced the toxicity and caspase-3 activation caused by expression of expanded polyglutamine tract. The outcome of a recent breakthrough in studying spinocerebella ataxia 1 (SCA1) demonstrated a bright future in using RNAi in HD[52]. Both SCA1 and HD belong to a group of severe neurodegenerative diseases caused by expansion of the polyglutamine tract in proteins. In the study, after recombinant adeno-associate virus (AAV) vectors expressing short-hairpin RNA (shRNA) were injected into the cerebellar region of the brain of transgenic SCA1 mice expressing transgenic human disease allele (ataxin-1-Q82), the cerelellar morphology was significantly improved and ataxin-1 inclusions were reduced in Purkinje cells, while there was no effect of the RNAi vectors in wild-type mice[52]. RNAi targets both the mutant and wild-type allele, however, it does not completely eliminate the mRNA and protein synthesis. Huntingtin plays a critical role during development as shown by the fact that huntingtin knock-out mice are embryonic lethal. However, reducing huntingtin after birth and before the onset of the disease could have minor effects on its normal function. Also, reduction of mutant huntingtin should delay the onset of disease or disease progression. Taken together, RNAi is a potential therapy for HD and will be tested in transgenic mice soon. RNAi targeting mutant huntingtin allele is also currently under develop-ment.
Full table
Neuron protection and replacement therapy
The goal of neuroprotective therapy is to preserve vulnerable neurons, restore neuronal function or replace lost neurons. These therapies do not require a full understanding of the molecular mechanisms of HD. Thus, they could be useful strategies for the present treatment of HD.
Neurotrophic factors Neurotrophic factors interact with three types of membrane receptors, named TrKA, TrKB, and TrKC. These receptors have tyrosine kinase activity. Nerve growth factor (NGF) binds to TrKA receptors, and brain derived neurotrophic factor (BDNF) and neurotrophin-4/5 (NT-4/5) bind to TrkB, whereas neurotrophin-3 (NT-3) binds preferentially to TrkC[53]. Neurotrophic factors play critical roles in neurodevelopment and support neuronal survival and plasticity of mature neurons. In a variety of neuronal injury models, neurotrophic factors have been shown to protect neurons from death and to promote functional recovery. Thus, neurotrophic factors have long been suggested as therapeutic candidates for neurodegenerative disorders including Parkinson disease and HD.
Beneficial effects of neurotrophic factors in HD have been demonstrated in animal models of HD. Excitotoxins and mitochondrial toxins have been used for producing animal models of HD because they induce degeneration of striatal neurons mimicking pathological features of HD. In these HD animal models, administration of neurotrophic factors reduced neuronal damage and behavioral deficits. Neuro-trophins are introduced into brains by direct injection, implantation of engineered cells producing neurotrophins, or using viral expression vectors. Direct injection of neuro-trophins into brains would not be clinically practical, but transplantation of neurotrophin producing cells into brains is feasible to achieve long lasting delivery of neurotrophins[54]. To minimize immunoresponse to implanted cells, different methods of encapsulation of cells have been developed. These technologies have greatly reduced host immune response and prolonged survival of implanted cells. In a reported study, polymer-encapsulated cells secreting human NGF survived as long as one year after transplantation into rat ventricles[55].
Instriatal administration of quinolinic acid (QA), a selective NMDA receptor agonist, causes apoptotic death of striatal projection neurons, but striatal interneurons are mostly spared[56]. This pathology is similar to that found in HD brains. In QA-induced rodent models of HD, intrastriatal injection of adenovirus encoding BDNF protected neurons from QA-induced striatal lesions as revealed by histological examinations. Immunocytochemistry revealed that viral expression of BDNF increased survival of striatal GABAergic projection neurons[57]. In another study, cell lines expressing BDNF, NT-3, or NT-4/5 were grafted in the adult rat striatum before QA injection. Seven days after QA injection, immunohistological examination confirmed protection of striatal projection neurons by neurotrophins. QA injection alone or in combination with the control cell line induced a selective loss of striatal projection neurons. Grafting of a BDNF-secreting cell line prevented the loss of all types of striatal projection neurons analyzed[58]. Neuroprotective effects of transplantation of CTNF producing cells were also demonstrated in non-human primate HD models. Emerich et al gave Cynomolgus monkeys intrastriatal implants of polymer-encapsulated kidney fibroblasts from baby hamsters that had been genetically modified to secrete human CNTF[59]. Human CNTF was found to exert a neuroprotective effect on striatal neurons, including GABAergic, cholinergic and diaphorase-positive neurons. Human CNTF also prevented the retrograde atrophy of layer V neurons in the motor cortex and exerted a significant protective effect on the GABAergic innervation of the two important target fields of the striatum[59].
Another useful method for delivery of neurotrophins is to use a viral expression vector. Introduction of viral vector into brains can achieve local production of exogenous neurotrophic factors. In 3-nitropropionic acid (3-NP)-induced rodent HD model, Jodi et al investigated the neuroprotective effects of glial-derived neurotrophic factor (GDNF) delivered by an AAV. Lewis rats received bilateral injections of either AAV-GDNF or AAV-green fluorescence protein into the striatum followed by chronic subcutaneous infusions of the mitochondrial toxin, 3-NP (38 mg/kg). All rats underwent 4 weeks of behavioral testing and were then killed. Following 3-NP, the motor performance of AAV-GFP-treated rats on a raised platform deteriorated, while the performance by AAV-GDNF-treated rats was near normal. Histological analysis found that 3-NP-treated rats receiving AAV-GDNF displayed 70% more NeuN-immunoreactive neurons compared to 3-NP-treated rats receiving AAV-GFP[60]. Other studies on QA-induced rodent HD models also found that enhanced expression of BDNF or GDNF, achieved by AAV vector-mediated gene delivery, protected striatal neurons. Both AAV-BDNF and AAV-GDNF significantly reduced the loss of NeuN and calbindin-immunopositive striatal neurons 2 weeks after lesion compared to controls[61].
Neurotrophins not only reduced neuronal loss in chemical models of HD, but also improved behavioral deficits. In rats, bilateral infusion of QA produced a significant loss of bodyweight and mortality, which was prevented by prior implantation of hCNTF-secreting cells. QA produced impairments in motor function and cognitive function. The behavioral deficits induced by QA were abolished by implantation of hCNTF-secreting cells prior to QA infusion. Consistent with behavioral improvements after implantation of hCTNF secreting cells, neurochemical examination revealed that CNTF-treated animals did not exhibit any decrease in ChAT levels and only a 10% decrease in GAD levels in the striatum, whereas in control animals QA decreased striatal ChAT levels by 35% and striatal GAD levels by 45%[59]. Further studies in primates also showed that intracerebral delivery of low doses of CNTF at the onset of HD symptoms not only protects neurons from degeneration but also restores neostriatal functions[62]. These results support the hypothesis that CNTF infusion into the striatum of HD patients could not only block the degeneration of neurons but also alleviate motor and cognitive symptoms associated with persistent neuronal dysfunction.
The elucidation of the genetic defect in patients with HD has allowed for the detection of individuals at risk for HD prior to the onset of symptoms. Thus "neuroprotection strategies" aimed at preventing the neuropathological and behavioral sequelae of this disease might be powerful therapeu-tically, as they could be introduced to healthy patients before the initiation of a massive degenerative cascade principally localized to the striatum. After some issues, such as immunoresponse and long-term survival of transplanted cells, are resolved this type of therapeutic strategy could be ready for clinical testing in HD patients.
Neural cell transplantation For many years investigators have proposed the replacement of lost neurons and the restoration of functions by grafting new cells into specific brain regions. HD pathology affects multiple brain regions, but the striatum is most affected in the early stages. Now many studies have been conducted in animal models and HD patients, giving HD patients new hope for a potential novel therapy.
In early studies on a rodent HD model, intrastriatal injection of striatal neurons obtained from 14 to 15-day-old rat fetuses re-established a new striatum-like structure at the site of the ibotenic acid-induced lesions. This reduced striatal atrophy on average to approximately 50%-70% of normal control in the rats with lesions and to approximately 30%-40% in the animals with grafts. In the rats with grafts, there was a significant recovery of striatal neuronal markers after excitotoxic lesions[63]. Both cell suspensions and tissue chunks can be implanted into the striatum. But grafts derived from cell suspensions triturated in the presence of trypsin contained larger quantities of striatal tissue within the graft and more DARPP-32-positive medium spiny neurons than grafts implanted as fragments of tissue. Afferent and efferent connectivity was also better in the trypsinized suspension graft group[64].
The rationale for neuronal replacement as a possible therapy for HD is strengthened by showing functional recovery after cell transplantation in a primate HD model. A complete and persistent recovery was demonstrated in a frontal-type cognitive task 2 to 5 months after intrastriatal allografting. The striatal allografts also reduced the occurrence of dystonia, a major abnormal movement associated with HD[65]. These results further support the use of neural transplantation as a potential therapy for HD. Grafted cells must establish connection with target neurons to have their functions. The establishment of a neuronal network was revealed in the experiments with transplantation of neural precursor cells into a rat model of HD. Neural precursors survived transplantation and large numbers differentiated to express neuronal markers, including DARPP-32, indicating a mature striatal phenotype had been adopted. Neuronal fibers from the grafts projected diffusely throughout the host brain, although there was no evidence that outgrowth was specifically target directed[66]. The thalamostriatal projections to rat neostriatal grafts were studied using the Phaseolus vulgaris-leucoagglutinin (PHA-L) axonal tracing technique. Two to six months after implantation of striatal primordia into adult neostriata, PHA-L was injected into two different portions of the intralaminar nuclear complex of the thalamus and examined with an electron microscope. Some of the labeled fibers in the grafts formed dense, focal arborizations. However, the synaptic connections in grafts were different from the normal striatum. The shift of postsynaptic elements in the grafts suggests a loss of pathway specificity in the induction of dendritic spines on neostriatal neurons in grafts[67]. In a QA-induced experimental HD model, DA-mediated electrophysiological depression was studied after fetal striatal tissue transplants were grafted. QA lesioning reduced responses to DA in the striatal neurons. The dose of DA required to inhibit striatal neuron activity in the lesioned rats was significantly increased compared to that in the non-lesioned rats. Transplantation of the fetal striatal tissue restored electrophysiological sensitivity to DA in the lesioned striatum. Tyrosine hydroxylase-positive terminals were found innervating the striatal grafts[68]. The survival of grafts and the effects of the grafts on the development of neurological deficits in HD transgenic mice have also been examined. Hemizygote transgenic and wild-type littermate female mice received striatal grafts at the 10 weeks of age and were allowed to survive for 6 weeks. Normal healthy grafts were seen to survive and differentiate within the striatum of transgenic mice in a manner comparable to that seen in control mice. The transgenic mice exhibited a progressive decline in bodyweight from 9 weeks of age and a progressive hypoactivity in an open field test of general locomotor behavior. Although striatal grafts exerted a statistically significant influence on several indices of this impairment, all behavioral effects were small and did not exert any clinically relevant effect on the profound neurological deficiency of the transgenic mice[69]. This could be a result of the rapid development of neurological deficits in this HD model.
The safety and therapeutic effects of neural transplantation have been tested in HD patients. The first clinical trial in HD patients was reported in 1995 by Madrazo et al[70]. In their trial, homotopic fetal striatal homotransplantations were performed on two HD patients. Each patient was implanted in the ventricular wall of the right caudate nucleus with striata from a 13 week-old and a 12 week-old human fetus. After surgery both patients were kept on cyclosporine A for immunosuppression. The two patients were observed for 16-33 months post-transplantation for neurological progression of their disease. The results showed that the disease in both patients had progressed slower in relation to their preoperative state. The effects of fetal tissue transplantation on HD symptoms were carefully evaluated in three patients who had received surgery. Four to six months after surgery, all patients demonstrated increased scores on some measures of cognitive functions. These findings suggest that fetal striatal transplantation could improve some of the cognitive symptoms associated with HD[71].
In a safety evaluation of three HD patients who received fetal striatal tissue transplantation for one year, MRI revealed that the grafts survived and grew within the striatum without displacing the surrounding tissue. No patients demonstrated adverse effects from the surgery or the associated cyclosporin immunosuppression, nor did any patient exhibit deterioration following the procedure[72]. In a post-mortem examination of an HD patient, who died from cardiovascular disease 18 months after transplantation, histological analysis demonstrated surviving transplanted cells with typical morphology of the developing striatum. Selective markers of both striatal projection and interneurons showed positive transplant regions clearly innervated by host tyrosine hydroxylase fibers. Neuronal protein aggregates of mutant htt, a biohallmark of HD neuropathology, were not found within the transplanted fetal tissue[73]. These results suggest that grafts derived from human fetal striatal tissue can survive for a relatively long time after transplantation into a patient with HD[73]. Functional improvements were demonstrated in the Gaura et al report with three HD patients[74]. They showed that the clinical changes in these three patients were associated with a reduction of the striatal and cortical hypometabolism, demonstrating that grafts were able to restore the function of striato-cortical loops. Conversely, in the two patients not improved by the grafts, striatal and cortical hypometabolism progressed over the 2-year follow-up. Finally, detailed anatomical/functional analysis of the grafted striata, enabled by the 3D fusion of MRI and metabolic images, revealed considerable heterogeneity in the anatomic and metabolic profiles of grafted tissue, both within and between HD patients.
The safety of fetal striatal tissue transplantation was demonstrated in a few clinical trials[75-78]. All these studies reported that surgical procedures were safe and patients tolerated grafts well. The major adverse events related to the procedure were associated with immunotherapy. However, a report from Hauser et al raised big concerns on the safety of fetal tissue transplantation[79]. In their clinical trial of 7 patients undergoing bilateral engraftment, there were 4 subdural haematomas (in 3 subjects), 2 requiring neurosurgical drainage. One subject died 18 months after surgery from probable cardiac arrhythmia secondary to severe atherosclerotic cardiac disease. Autopsy demonstrated clearly demarcated grafts of typical developing striatal morphology, with host-derived dopaminergic fibers extending into the grafts and no evidence of immune rejection. These latter subjects might have had more advanced HD with a greater degree of brain atrophy, and this could be a contraindication to engraftment.
Other neuroprotective approaches Mitochondria dysfunction has been implicated in HD pathogenesis. The supporting evidence includes deficiency in mitochondria complex I, II and IV in HD brains and HD transgenic mice. Toxins inhibiting mitochondria complex produce striatal HD pathology. Therefore, compounds enhancing energy metabolism have been evaluated for treatment of HD. In HD patients, lactate was increased in cerebrospinal fluid and cerebral cortex. Treatment with coenzyme Q10 (CoQ10), an essential cofactor of the electron transport chain and an important antioxidant, resulted in significant decreases in cortical lactate concentrations in 18 patients. These findings provide evidence for a generalized energy defect in HD, and suggest a possible therapy[54]. Thus, its neuroprotective effects have been investigated in a variety of animal models. These studies have demonstrated that CoQ10 can protect against striatal lesions produced by the mitochondrial toxins malonate and 3-nitropropionic acid. Oral administration of CoQ10 significantly decreased elevated lactate levels in patients with HD[80]. CoQ10 in combination with the NMDA antagonist remacemide was examined to determine the efficacy in ameliorating the motor dysfunction and premature death of HD transgenic mice. Motor performance was specifically but transiently improved beginning 3 weeks after drug adminis-tration. Survival, however, was not prolonged[81]. In another study in HD transgenic mice with CoQ10 and remace-mide, Ferrante et al found that oral administration of either CoQ10 or remacemide significantly extended survival and delayed the development of motor deficits, weight loss, cerebral atrophy, and neuronal intranuclear inclusions in the R6/2 transgenic mouse model of HD. The combined treatment, using CoQ10 and remacemide, was more efficacious than either compound alone, resulting in a 32% and 17% increase in survival in the R6/2 and N171-82Q mice, respectively[82].
Huntington et al completed a multicenter, parallel group, double-blind, 2×2 factorial, randomized clinical trial with CoQ10 in 2001[83]. Research participants with early HD (n=347) were randomized to receive CoQ10 300 mg bid, remacemide hydrochloride 200 mg three times daily, both, or neither treatment, and were evaluated every 4 to 5 months for a total of 30 months on assigned treatment. The pres-pecified primary measure of efficacy was the change in total functional capacity (TFC) between baseline and 30 months. This study, however, yielded limited beneficial results. Neither intervention significantly altered the decline in TFC. Patients treated with CoQ10 showed a trend toward slowing in TFC decline (13%) over 30 months as well as beneficial trends in some secondary measures. There were increased frequency of nausea, vomiting, and dizziness with remace-mide, and an increased frequency of stomach upset with CoQ10.
Another compound having an effect on bioenergy metabolism is creatine. In HD-transgeneic mice, dietary supplementation of 2% creatine significantly improved survival, slowed the development of motor symptoms, and delayed the onset of weight loss. Creatine lessened brain atrophy and the formation of intranuclear inclusions[84]. Later studies suggested that creatine treatment started after onset of clinical symptoms in HD mice was able to significantly extend survival and improve motor performance. There was reduced brain levels in both creatine and ATP in R6/2 mice, consistent with a bioenergetic defect. Oral creatine supplementation significantly increased brain concentrations of creatine and ATP to wild-type control level[85]. These studies suggest that creatine might be useful in the treatment of HD. A preliminary clinical trial with creatine has been performed in 41 patients for 1 year. Creatine 5 g/d was admini-stered. At baseline and after 6 and 12 months, the functional, neuromuscular, and cognitive status of the patients was assessed by a test battery that consisted of: (1) the Unified Huntington's Disease Rating Scale (UHDRS); (2) an exercise test on an isokinetic dynamometer to assess strength of the elbow flexor muscles; (3) a maximal exercise test on a bicycle ergometer to evaluate cardiorespiratory fitness; and (4) a test to assess bimanual coordination ability. One year of creatine intake, at a rate that can improve muscle functional capacity in healthy subjects and patients with neuromuscular disease (5 g/d), did not improve functional, neuro-muscular, and cognitive status in patients with stage I to III HD[86].
It has been suggested that excitotoxicity contributes to the pathogenesis of HD. Riluzole is a substance with glutamate antagonistic properties that is used for neuropro-tective treatment in amyotrophic lateral sclerosis, and which is currently being tested in clinical trials for treatment of HD. R6/2 transgenic mice were treated with riluzole orally beginning at a presymptomatic stage until death to investigate its potential neuroprotective effects in the mouse model and it was found that survival time in the riluzole group was significantly increased in comparison to placebo-treated trans-genic controls. Additionally, the progressive weight loss was delayed and significantly reduced by riluzole treatment; behavioral testing of motor coordination and spontaneous locomotor activity, however, showed no statistically significant differences. These data suggest that riluzole is a promising candidate for neuroprotective treatment in human HD[11].
Recently a few studies have produced interesting results on the beneficial effects of environment enrichment on HD. van Dellen et al housed HD mice in large standard cages that contained cardboard, paper and plastic objects, which were changed every two days, from the age of 4 weeks[87]. They showed that exposure of these mice to a stimulating, enriched environment from an early age helps to prevent the loss of cerebral volume and delays the onset of motor disorders. In the brains of humans diagnosed with HD cannabinoid CB1 receptors are selectively lost from the basal ganglia output nuclei prior to the development of other identifiable neuropathology. In animal studies, they further showed that HD mice housed in a normal environment show a loss of cannabinoid CB1 and dopamine D1 and D2 receptors in the striatum and the corresponding output nuclei of the basal ganglia. HD mice exposed to an enriched environment showed equivalent loss of D1 and D2 receptors as their "non-enriched"? counterparts; in contrast, the "enriched"? mice show significantly less depletion of CB1 receptors. These results therefore show that an enhanced environment slows the rate of loss of one of the first identifiable neurochemical deficits of HD[88]. Environment enrichment slowed a decline in RotaRod performance in R6/2 mice. Enrichment also delayed the loss of peristriatal cerebral volume in R6/2 brains[89]. Improvement of motor functions and prevention of loss of bodyweight in HD mice were found. HD patients and transgenic mice expressing mutant human huntingtin exhibit reduced levels of BDNF, hyperglycemia, and tissue wasting. Enrichment entirely corrected these changes. BDNF levels are unaltered in HD in the anterior cortex, suggesting that enrichment might prevent HD-induced impairment of anterograde transport of this neurotrophin to the striatum[90].
It has been shown that dietary restriction (DR) slows down the aging process and increases life expectancy. Further, the progression of neuropathological (formation of huntingtin inclusions and apoptotic protease activation), behavioral (motor dysfunction), and metabolic (glucose intolerance and tissue wasting) abnormalities in HD mice were shown to be retarded when the mice were maintained on a DR feeding regimen, which resulted in an extension of their lifespan. DR increases levels of brain-derived neurotrophic factor and the protein chaperone heat-shock protein-70 in the striatum and cortex, which are depleted in HD mice fed a normal diet. These results suggest a dietary intervention that could suppress the disease process and increase the lifespan of humans that carry the mutant huntingtin gene[91]. These results could provide a molecular basis for ameliorating the effects of HD by some simple yet effective appro-aches.
Symptomatic treatment
Movement disorder is a prominent feature of HD. The motor component of HD consists of involuntary choreiform movements and increasing difficulties with voluntary movement. Behavioral and psychological problems are the first manifested symptoms in nearly 50% of HD patients. The most common symptoms are loss of energy and initiative, poor perseverance and quality of work, impaired judgment, poor self-care, and emotional blunting. Affective symptoms such as depression, anxiety, irritability, dementia, and schizophrenia-like psychosis occur with a relatively high frequency in HD[92,93]. At present, in the absence of a cure for HD, it is desirable to treat HD symptoms aimed at enhancing the quality of life of patients and improving overall functioning. These types of treatments should include medication, social service, and physical/occupational therapy.
Management of motor dysfunction In general, treatment of chorea is not recommended unless symptoms are disabling. Degeneration of striatal GABAergic neurons can cause hyperactivity of dopaminergic systems. Reducing dopaminergic activity might help restore the balance of neurotransmitter functions. Neuroleptics are dopamine receptor antagonists used for the treatment of psychosis. These drugs have been tried for treatment of choreiform movement in HD patients. Haloperidol is a protoform neuroleptic for the treatment of schizophrenia. Haloperidol is a dopamine D2 receptor antagonist. This drug has been tested in HD for reducing abnormal movements. Eighteen patients with Huntington's chorea were examined before and after treatment with haloperidol, pimozide and tiapride to study the effect of such treatment on hyperkinesia and motor perfor-mance. Pimozide and haloperidol improved hyperkinesia; none of the drugs significantly affected motor performance[94]. The relationship between serum haloperidol concentration and improvement in abnormal movements was investigated in 20 adult HD patients. Serum samples and assessments of the severity of chorea were simultaneously obtained from each patient. Results showed that significant improvement of abnormal movements, greater than 30% from baseline, occurred at serum concentrations between 2 and 5 µg/L, which corresponded to doses of 1.5 to 10 mg/d[95]. The main adverse effect of haloperidol is the high frequency of dyskinesia. Clinically the most problematic of these are sedation, cognitive slowing, increased mobility problems, and hypotension. The inability of traditional dopamine antagonists to improve functional capacity, despite ameliorating chorea, is possibly a result of the suppression of voluntary motor activity. The antipsychotic drug risperidone was reported to be effective in the treatment of chorea and psychosis in HD patients. After initiating the treatment, the patient experienced improvement in maintaining gait and increased hand skills. It was noticed that the patient had decreased choreiform movements in the second week of the treatment[96].
Chorea in HD and in the levodopa-induced dyskinesias of PD can be clinically indistinguishable. In PD, hyperphos-phorylation of NMDA receptors expressed on striatal medium spiny neurons contributes to peak-dose dyskinesias, and drugs that block these receptors can diminish chorea severity. Because these spiny neurons are the primary target of the neurodegenerative process in HD, sensitization of NMDA receptors on residual striatal neurons might also participate in the generation of motor dysfunction in HD. Movement disorder is also treated with the NMDA receptor antagonists riluzole and amantadine. In a 6-week open-label trial of riluzole (50 mg twice a day) in eight subjects with HD, subjects were evaluated before riluzole treatment, on treatment, and off treatment, with the chorea, dystonia, and total functional capacity (TFC) scores from the Unified Huntington's Disease Rating Scale, and magnetic resonance spectroscopy measurements of occipital cortex and basal ganglia lactate levels. The chorea rating score improved by 35% on treatment and worsened after discontinuation of treatment. There was no significant effect on the dystonia or TFC score[97]. In another open label study, riluzole was given (50 mg twice a day) to nine HD patients (clinical stages 1-3; mean age 46.4 years; mean disease duration 8 years). Patients were evaluated at baseline and after 3 and 12 months of riluzole therapy. Results demonstrated that riluzole was well tolerated. At 3 months, mean total motor scale (TMS), mean TMS chorea subscore, and mean total functional capacity scale were significantly improved compared with pre-drug baseline. At 12 months, however, this beneficial effect on motor status and overall function was not sustained. In contrast, severity and frequency of behavioral dysfunction as well as psychomotor speed assessed by the symbol digit modalities test were improved compared with baseline. These data suggest that there are transient antichoreatic effects and more sustained effects of riluzole on psychomotor speed and behavior in patients with HD[98]. In a case report presented by Bonelli et al, two HD patients with severe motor dysfunction were initially treated with olanzapine (20 mg daily)[99]. Improvement of motor performance was obtained with olanzapine in both patients. After 2 weeks of treatment with olanzapine, riluzole (50 mg twice a day) was added. A further slight improvement of motor function (especially in the fine motor tasks) was seen in the next 2 weeks without additional side-effects.
Another NMDA receptor antagonist tested for use in HD patients is amantadine. In a clinical study conducted by Verhagen et al, 24 patients with HD entered a double-blind placebo-controlled crossover study of amantadine with two 2-week arms[100]. Chorea scores were lower with amantadine (usually 400 mg/d) than placebo, with a median reduction in extremity chorea at rest of 36% for all 22 patients evaluated and of 56% in the 10 individuals with the highest plasma drug levels. Improvement correlated with plasma amantadine concentrations but not CAG repeat length. The acute antidyskinetic effects of IV amantadine in HD were evaluated. A 2-h IV infusion of amantadine or placebo was given to 9 patients with HD on two different days in a double-blind, randomized crossover fashion. All patients subsequently received oral amantadine unblinded for a 1-year period. A reduction of dyskinesia scores was reported during both IV and oral amantadine treatment (P<0.05)[101].
Management of psychological disturbance There are not many studies on the treatment of the psychopathology of HD although psychological disturbance is common in HD patients. Episodic aggressive behavior in HD that responded poorly to neuroleptics was reduced by a carefully titrated dose of propranolol in 3 patients with advanced HD. The optimal doses were 180 and 30 mg/d, respectively[102]. Lithium is a mood stabilizer used for bipolar disorder. It has been reported that 6 patients with a family history of HD chorea participated in a double blind crossover trial involving four treatments: lithium carbonate, haloperidol, lithium carbonate and haloperidol, and placebo. None of the treatments significantly affected chorea measurements. With regard to the psychological variables, the levels of irritability, the frequency of angry outbursts and depression did appear to be affected by the treatment in some patients. Three patients improved on a combination of lithium carbonate and haloperidol while the remaining three did not. It is suggested that lithium carbonate and haloperidol together should be seriously considered in the treatment of HD when patients are excessively irritable and impulsive[103].
Atypical antipsychotic drugs have recently been found to be useful in the treatment of HD symptoms including motor dysfunction. Risperidone is a novel antipsychotic drug with a balanced serotonin 5-HT2 and dopamine D2 receptor antagonism. In a case report, risperidone was introduced in combination with clozapine therapy. Risperidone was started at 0.5 mg/d and gradually increased to 6 mg/d over a 6-week period. This coincided with marked improvement in abnormal involuntary movements (AIMS score of 7) and further improvement in psychosis (BPRS score of 7). Choreiform movements of the limbs ceased almost completely, gait improved significantly, and dysarthria and facial grimacing decreased[104]. Olanzapine is a new atypical antipsychotic drug. It is a thienodibenzodiazepine and is structurally very similar to clozapine. Olanzapine has been shown to have a high affinity for a large number of receptors including D1, D2, D4, 5HT2A, 5HT2C, 5HT3, α1-adrenergic, histamine H1, and five muscarinic receptors. Unlike clozapine it is not associated with the potentially serious side effect of agranulocytosis and therefore frequent blood monitoring is not necessary. Dipple reported the first case of using olanzapine in HD[105]. An HD patient who had shown no improvement after using other typical anti-psychotic drugs showed a positive response in improving motor function with olanzapine. As a result, more HD patients were treated with olazapine. In a clinical study with 11 HD patients, 9 patients were treated with olanzapine for 9.8-5.9 months. Assessment of the drug's effects was carried out using the Clinical Global Impression of Change Scale (CGIC) and the United Huntington's Disease Rating Scale behavioral (UHDRS-b) and motor (UHDRS-m) at 6-month intervals. The Mean CGIC was 2.1?0.8. UHDRS-b improved significantly and UHDRS-m did not change. It was concluded that olanzapine is a good alternative treatment in HD, mainly for psychiatric symptoms and is moderately effective for motor symptoms, possibly because of its effect on chorea[106].
Symptomatic treatment of HD like symptoms with Chinese medicine HD diagnosis is difficult in China as autopsy and genetic testing are usually not performed. However, there are many reports describing successful treatment of choreiform movement disorders (named chorea minor) with Chinese medicine. Among these reported cases, most involve HD-like symptoms caused by other diseases. But these treatments might also be effective in relief of HD symptoms. Recent studies reported that a natural product derived from six herbs, including Ginsheng and Shouwu, protected 3-nitropropionic acid induced striatal lesions and reduced behavioral deficits. This herbal product increased expression of BDNF and GDNF in the brain[107-109].
Summary and conclusion
The therapeutic benefits of current symptomatic treatments remain to be determined. The effectiveness of drug treatment usually has been evaluated only in a small number of patients, and varies in patients with different stages of HD. In order to confirm the value of these treatments in the management of symptoms of HD, more clinical trials involving a larger number of patients and better design are needed. It should be noted that improvement of chorea by typical neuroleptics might be at the expense of worsening voluntary movement. Extensive studies have greatly advanced our understanding of the molecular mechanisms of HD pathogenesis. The main challenge in finding a cure for HD is the selection of molecular targets. Multiple parallel and sequential signaling pathways are involved in cultivating ultimate neuronal dysfunction and death induced by mutant huntingtin. Identifying upstream and core molecular events could be crucial for therapeutic development. Thus far, these key issues have not been resolved. Another big challenge is an effective read-out for the assay of therapeutic effects. Many novel therapies targeting molecular pathogenesis are awaiting safe and clinical studies. With regards to fetal tissue transplantation, the resources are limited and there is still debate on the ethical issues of using this technology. The safety and rationale of tissue transplantation have also now been questioned[110]. Compounds improving energy production are safe but their effectiveness needs to be further evaluated. At present, NMDA receptor antagonists, riluzole and amantadine, and energy production boosters, CoQ10 and creatine, might be worth testing. The use of Chinese medicine for HD treatment also needs further investiga-tion. We have made great progress, but we still have long way to go in developing a cure for HD.
Footnote
Supported by the National Natural Science Foundation of China (N
References
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971-83.
- Qin ZH, Gu ZL. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol Sin 2004;25:1243-9.
- Hughes RE, Olson JM. Therapeutic opportunities in polyglutamine disease. Nature Med 2001;7:419-23.
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997;90:549-58.
- Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, et al. Amyloid, formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somatic Cell Mol Gen 1998;24:217-33.
- Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington抯 disease pathology. Proc Natl Acad Sci USA 1999;96:4604-9.
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997;90:537-48.
- Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, et al. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington抯 disease therapy. Proc Natl Acad Sci USA 2000;97:6739-44.
- Pollitt SK, Pallos J, Shao J, Desai UA, Ma AA, Thompson LM, et al. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 2003;40:685-94.
- Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, et al. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington抯 disease by using an automated filter retardation assay. Proc Natl Acad Sci USA 2002;99:16400-6.
- Schiefer J, Landwehrmeyer GB, Luesse HG, Sprunken A, Puls C, Milkereit A, et al. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington抯 disease. Mov Disord 2002;17:748-57.
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004;10:148-54.
- Wang J, MacDonald ME, Gusella JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 2005. in press.
- Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003;421:373-9.
- Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington抯 disease. Neurobiol Dis 2004;16:546-55.
- Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington抯 disease. Human Mol Genet 2001;10:1307-15.
- Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington抯 disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol Cell 1998;1:595-601.
- Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, Becher MW, et al. Transglutaminase aggregates huntingtin into nonamyl-oidogenic polymers, and its enzymatic activity increases in Huntington抯 disease brain nuclei. Proc Natl Acad Sci USA 1999;96:7388-93.
- Lorand L, Parameswaran KN, Stenberg P, Tong YS, Velasco PT, Jonsson NA, et al. Specificity of guinea pig liver transglutaminase for amine substrates. Biochemistry 1979;18:1756-65.
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, et al. Therapeutic effects of cystamine in a murine model of Huntington抯 disease. J Neurosci 2002;22:8942-50.
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 2002;8:143-9.
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, et al. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Gen 1996;13:442-9.
- Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem 2000;275:19831-8.
- Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpaindependent proteolysis. Proc Natl Acad Sci USA 2001;98:12784-9.
- Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem 2004;279:20211-20.
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 2002;10:259-69.
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997;277:1990-3.
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, et al. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999;23:181-92.
- Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, et al. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci 1999;19:964-73.
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000;6:797-801.
- Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington抯 disease. Proc Natl Acad Sci USA 2003;100:10483-7.
- Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander RM, et al. Cytochrome C and caspase-9 expression in Huntington's disease. Neuromolecular Med 2002;1:183-95.
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 1999;399:263-7.
- Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 1999;22:623-33.
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 2001;293:493-8.
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 2002;22:1277-87.
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Gen 2004;20:146-54.
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Human Mol Gen 2000;9:1259-71.
- Chan EY, Luthi-Carter R, Strand A, Solano SM, Hanson SA, DeJohn MM, et al. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington's disease. Human Mol Gen 2002;11:1939-51.
- Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Human Mol Gen 2002;11:905-14.
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington抯 disease. Science 2002;296:2238-43.
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001;413:739-43.
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 2003;100:2041-6.
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 2004;13:1183-92.
- Alvarez RD, Barnes MN, Gomez-Navarro J, Wang M, Strong TV, Arafat W, et al. A cancer gene therapy approach utilizing an anti-erbB-2 single-chain antibody-encoding adenovirus (AD21): a phase I trial. Clin Cancer Res 2000;6:3081-7.
- Marasco WA, Haseltine WA, Chen SY. Design, intracellular expression, and activity of a human anti-human immunodeficiency virus type 1 gp120 single-chain antibody. Proc Natl Acad Sci USA 1993;90:7889-93.
- Lecerf JM, Shirley TL, Zhu Q, Kazantsev A, Amersdorfer P, Housman DE, et al. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Hunting-ton's disease. Proc Natl Acad Sci USA 2001;98:4764-9.
- Colby DW, Garg P, Holden T, Chao G, Webster JM, Messer A, et al. Development of a human light chain variable domain (V(L)) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J Mol Biol 2004;342:901-12.
- Khoshnan A, Ko J, Patterson PH. Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc Natl Acad Sci USA 2002;99:1002-7.
- Xia H, Mao Q, Paulson HL, Davidson BL. siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol 2002;20:1006-10.
- Caplen NJ, Taylor JP, Statham VS, Tanaka F, Fire A, Morgan RA. Rescue of polyglutamine-mediated cytotoxicity by double-stranded RNA-mediated RNA interference. Hum Mol Gen 2002;11:175-84.
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 2004;10:816-20.
- Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol 1994;25:1386-403.
- Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann Neurol 1997;41:160-5.
- Winn SR, Lindner MD, Lee A, Haggett G, Francis JM, Emerich DF. Polymer-encapsulated genetically modified cells continue to secrete human nerve growth factor for over one year in rat ventricles: behavioral and anatomical consequences. Exp Neurol 1996;40:126-38.
- Qin ZH, Wang Y, Chase TN. Stimulation of NMDA receptors induces apoptosis in rat brain. Brain Res 1996;725:166-76.
- Bemelmans AP, Horellou P, Pradier L, Brunet I, Colin P, Mallet J. Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington's disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther 1999;10:2987-97.
- Perez-Navarro F, Canudas AM, Akerud P, Alberch J, Arenas E. Brain-derived neurotrophic factor, neurotrophin-3, and neuro-trophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington's disease. J Neurochem 2000;75:2190-9.
- Emerich DF, Winn SR, Hantraye PM, Peschanski M, Chen EY, Chu Y, et al. Protective effect of encapsulated cells producing neurotrophic factor CNTF in a monkey model of Huntington's disease. Nature 1997;386:395-9.
- McBride JL, During MJ, Wuu J, Chen EY, Leurgans SE, Kordowera JH. Structural and functional neuroprotection in a rat model of Huntington's disease by viral gene transfer of GDNF. Exp Neurol 2003;181:213-23.
- Kells AP, Fong DM, Dragunow M, During MJ, Young D, Connor B. AAV-mediated gene delivery of BDNF or GDNF is neuro-protective in a model of Huntington disease. Mol Therapy 2004;9:682-8.
- Mittoux V, Joseph JM, Conde F, Palfi S, Dautry C, Poyot T, et al. Restoration of cognitive and motor functions by ciliary neurotrophic factor in a primate model of Huntington抯 disease. Hum Gene Ther 2000;20:1177-87.
- Isacson O, Brundin P, Gage FH, Bjorklund A. Neural grafting in a rat model of Huntington's disease: progressive neurochemical changes after neostriatal ibotenate lesions and striatal tissue grafting. Neuroscience 1985;16:799-817.
- Watts C, Brasted PJ, Dunnett SB. The morphology, integration, and functional efficacy of striatal grafts differ between cell suspensions and tissue pieces. Cell Transplant 2000;9:395-407.
- Palfi S, Conde F, Riche D, Brouillet E, Dautry C, Mittoux V, et al. Fetal striatal allografts reverse cognitive deficits in a primate model of Huntington disease. Nat Med 1998;4:963-6.
- Armstrong RJ, Watts C, Svendsen CN, Dunnett SB, Rosser AE. Survival, neuronal differentiation, and fiber outgrowth of propagated human neural precursor grafts in an animal model of Huntington's disease. Cell Transplant 2000;9:55-64.
- Xu ZC, Wilson CJ, Emson PC. Restoration of thalamostriatal projections in rat neostriatal grafts: an electron microscopic analysis. J Comp Neurol 1991;303:22-34.
- Chena GJ, Jengb CH, Lina SZ, Tsaic SH, Wangd Y, Chianga YH. Fetal striatal transplants restore electrophysiological sensitivity to dopamine in the lesioned striatum of rats with experimental Huntington's disease. J Biomed Sci 2002;9:303-10.
- Dunnett SB, Carter RJ, Watts C, Torres EM, Mahal A, Mangiarini L, et al. Striatal transplantation in a transgenic mouse model of Huntington's disease. Exp Neurol 1998;154:31-40.
- Madrazo I, Franco-Bour RE, Castrejon H, Cuevas C, Ostrosky-Solis F. Fetal striatal homotransplantation for Huntington's disease: first two case reports. Neurol Res 1995;17:312-5.
- Philpott LM, Kopyov OV, Lee AJ, Jacques S, Duma CM, Caine S, et al. Neuropsychological functioning following fetal striatal transplantation in Huntington抯 chorea: three case presentations. Cell Transplant 1997;6:203-12.
- Kopyov OV, Jacques S, Lieberman A, Duma CM, Eagle KS. Safety of intrastriatal neurotransplantation for Huntington抯 disease patients. Exp Neurol 1998;49:97-108.
- Freemana TB, Cicchettid F, Hauserb RA, Deaconf TW, Li XJ, Herschi SM, et al. Transplanted fetal striatum in Huntington抯 disease: phenotypic development and lack of pathology. Proc Natl Acad Sci USA 2000;97:13877-82.
- Gaura VR, Bachoud-Levi AC, Ribeiro MJ, Nguyen JP, Frouin V, Baudic S, et al. Striatal neural grafting improves cortical metabolism in Huntington's disease patients. Brain 2004;127:65-72.
- Bachoud-Levi AC, Nguyen JP, Brugieres P, Lefaucheur JP, Cesaro P, P, Remy P, et al. Motor and cognitive improvements in patients with Huntington's disease after neural transplantation. Lancet 2000;356:1975-9.
- Bachoud-Levi AC, Bourdet C, Brugieres P, Nguyen JP, Grand-mougin T, Haddad B, et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington's disease. Exp Neurol 2000;161:194-202.
- Fink JS, Schumacher JM, Ellias SL, Palmer EP, Saint-Hilaire M, Shannon K, et al. Porcine xenografts in Parkinson抯 disease and Huntington's disease patients: preliminary results. Cell Transplant 2000;9:273-8.
- Rosser AE, Barker RA, Harrower T, Watts C, Farrington M, Ho AK, et al. Unilateral transplantation of human primary fetal tissue in four patients with Huntington's disease: NEST-UK safety report. J Neurol Neurosurg Psychiatry 2002;73:678-85.
- Hauser RA, Furtado S, Cimino CR, Delgado H, Eichler S, Schwartz S, et al. Bilateral human fetal striatal transplantation in Huntington's disease. Neurology 2002;58:687-95.
- Beal MF. Coenzyme Q10 administration and its potential for treatment of neurodegenerative diseases. BioFactors 1999;9:261-6.
- Schillinga G, Coonfielda ML, Rossb CA, Borchelta DR. Coenzyme Q10 and remacemide hydrochloride ameliorate motor deficits in a Huntington抯 disease transgenic mouse model. Neurosci Lett 2001;315:149-53.
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante AL, Jenkins BG, Hersch SM, et al. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci 2002;22:1592-9.
- Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology 2001;57:397-404.
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Homas M, et al. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Disease 2001;8:479-91.
- Dedeoglu A, Kubilus JK, Yang L, Ferrante KL, Hersch SM, Beal MF, et al. Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington's disease transgenic mice. J Neurochem 2003;85:1359-67.
- Verbessem P, Lemiere J, Eijnde BO, Swinnen S, Vanhees L, van Leemputte M, et al. Creatine supplementation in Huntington's disease: a placebo-controlled pilot trial. Neurology 2003;61:925-30.
- van Dellen A, Blakemore C, Deacon R, York D, Hannan AJ. Delaying the onset of Huntington's in mice. Nature 2000;404:721-2.
- Glass M, van Dellen A, Blakemore CC, Hannand CAJ, Faullb RLM. Delayed onset of Huntington's disease in mice in an enriched environment correlates with delayed loss of cannabinoid CB1 receptors. Neuroscience 2004;123:207-12.
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, et al. Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol 2002;51:235-42.
- Spires TL, Grote HE, Varshney NK, Cordery PM, van Dellen A, Blakemore C, et al. Environmental enrichment rescues protein deficits in a mouse model of Huntington抯 disease, indicating a possible disease mechanism. J Neurosci 2004;24:2270-6.
- Duan W, Guo Z, Jiang H, Ware M, Li X-J, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA 2003;100:2911-6.
- Leroi I, Michalon M. Treatment of the psychiatric manifestations of Huntington's disease: a review of the literature. Can J Psychiatry 1998;43:933-40.
- Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol 2001;14:219-26.
- Girotti F, Carella F, Scigliano G, Grassi MP, Soliveri P, Giovannini P, et al. Effect of neuroleptic treatment on involuntary movements and motor performances in Huntington's disease. J Neurol Neurosurg Psychiatry 1984;47:848-52.
- Barr AN, Fischer JH, Koller WC, Spunt AL, Singhal A. Serum haloperidol concentration and choreiform movements in Huntington's disease. Neurology 1988;38:84-8.
- Erdemoglua AK, Boratavb C. Risperidone in chorea and psychosis of Huntington抯 disease. Eur J Neurol 2001;9:177-85.
- Rosas HD, Koroshetz WJ, Jenkins BG, Chen YI, Hayden DL, Beal MF, et al. Riluzole therapy in Huntington's disease (HD). Mov Disorders 1999;14:326-30.
- Seppi K, Mueller J, Bodner T, Brandauer E, Benke T, Weirich-Schwaiger H, et al. Riluzole in Huntington's disease (HD): an open label study with one year follow up. J Neurol 2001;248:866-9.
- Bonelli RM, Niederwieser G, Diez J, Tringer PK. Riluzole and olanzapine in Huntington抯 disease. Eur J Neurol 2001;9:177-85.
- Verhagen Metman L, Morris MJ, Farmer C, Gillespie M, Mosby K, Wuu J, et al. Huntington's disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002;59:694-9.
- Lucetti C, Del Dotto P, Gambaccini G. IV amantadine improves chorea in Huntington抯 disease: an acute randomized, controlled study. Neurology 2003;60:1995-7.
- Stewart JT, Mounts ML, Clark RL Jr. Aggressive behavior in Huntington抯 disease: treatment with propranolol. J Clin Psychiatry 1987;48:106-8.
- Leonard DP, Kidson MA, Brown JG, Shannon PJ, Taryan S. A double blind trial of lithium carbonate and haloperidol in Huntington's chorea. Aust N Z J Psychiatry 1975;9:115-8.
- Parsa MA, Szigethy E, Voci JM, Meltzer HY. Risperidone in treatment of choreoathetosis of Huntington's disease. J Clin Psychopharmacol 1997;17:134-5.
- Dipple HC. The use of olanzapine for movement disorder in Huntington's disease: a first case report. J Neurol Neurosurg Psychiatry 1999;67:123-4.
- Paleacu D, Anca M, Giladi N. Olanzapine in Huntington's disease. Acta Neurol Scand 2002;105:441-4.
- Zhang L, Zhang RY, Li L, Wang H, Xia J. Effect of Chinese compound 962 on behavior deficit in Huntington's disease model rats. Chin J Behav Med Sci 2002;3:243-6. Chinese.
- Zhang RY, Zhang L, Wang H, Yei CF. Effects of Shen-wu capsule on motor function andcontent of dopamine in striatum in Huntington model rats. Chin J Rehabil 2002;9:513-5. Chinese.
- Zhang RY, Li L, Zhang L, Wang H, Yei CF. Effects of Shenwu capsule on the behavior and neurotrophic factors in the rats with mimic Huntington's disease. J Beijing University TCM 2003;26:34-8.
- Albin RL. Fetal striatal transplantation in Huntington抯 disease: time for a pause. J Neurol Neurosurg Psychiatry 2002;73:612. [editorial commentaries: huntington's disease].