
Effect of pravastatin on impaired endothelium-dependent relaxation induced by lysophosphatidylcholine in rat aorta1
Introduction
Endothelial dysfunction is an early step in the pathogenesis of atherosclerosis and is characterized by an impaired endothelium function associated with a decreased production of nitric oxide (NO), reduced vasodilatation or abnormal vasoconstriction in response to acetylcholine. Oxidized low-density lipoprotein (ox-LDL) is an oxidative product of native LDL in vivo. It has been well established that ox-LDL plays an important role in the development of atherosclerosis[1]. Lysophosphatidylcholine (LPC) is the major component of ox-LDL[2–3]. LPC has been shown to increase the production of superoxide anions by activation of protein kinase C (PKC)[4] and has been implicated in impaired endothelium-dependent relaxation[2,3]. Exposure of normal blood vessels to LPC in vitro also mimicked the inhibitory effects of ox-LDL on endothelium-dependent relaxation[2,3]. Therefore, preventing the deleterious effects of LPC on endothelium is an effective approach for preventing the development of atherosclerosis.
Statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are cholesterol-lowering drugs and widely used in the treatment of hypercholesterolemia to prevent the development of atherosclerosis[5,6]. However, many studies have shown that statins have some beneficial effects on the cardiovascular system-independent of their cholesterol-lowering effect, such as restoring the endothelium-dependent relaxation function[7,8], preventing the adhesion of monocytes to endothelial cells[8], inhibiting the aggregation of platelets[9], suppressing the proliferation of vascular smooth muscle cells[10], and regulating angiogenesis[11] and blood pressure[12], which may be related to the up-regulation of nitric oxide synthase (NOS) expression. In addition, statins have been demonstrated to have anti-oxidative properties, as shown by their ability to reduce LDL oxidation[13] and scavenge oxygen-derived free radicals[14]. Pravastatin is a potent inhibitor of HMG-CoA reductase. Recently, a number of studies have demonstrated that pravastatin significantly inhibits the production of superoxide anions stimulated by phorbol ester in vascular endothelial cells[14], suppresses the increase in oxidative stress induced by LPC in vascular smooth muscle cells[15], and improves the impairment of endothelium-dependent relaxation in humans and animals with atherosclerosis and diabetes[16–18]. Therefore, it is important to determine whether pravastatin protects the vascular endothelium against damage due to LPC. In the present study, we sought to investigate the effects of pravastatin and its mechanisms on impaired endothelium-dependent relaxation induced by LPC in isolated rat aortic rings.
Materials and methods
Chemicals All chemicals were of the highest purity available. LPC, phenylephrine, acetylcholine, sodium nitroprusside, L-arginine, NG-nitro-L-arginine methyl ester (L-NAME), indomethacin, and chelerythrine were purchased from Sigma Chemical Co (St Louis, MO, USA). Superoxide dismutase (SOD) was the product of Changsha Biological Pharmaceutical Factory (Hu-nan, China). Pravastatin was kindly presented by Blue Treasure Pharmaceutical Factory (Guangdong, China). They were dissolved in distilled water and diluted with Krebs’ buffer before use.
Organ chamber experiments The study protocol was approved by the Animal Care and Use Committee of Central South University. Male Sprague-Dawley rats weighing 200-220 g were used. Rats were anaesthetized by intraperitoneal injection of sodium pentobarbital (30 mg/kg). The thoracic aorta was immediately isolated and placed in 4 ºC Krebs’ bicarbonate buffer of the following composition (in mmol/L): 118.3 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.0 NaHCO3, 11.0 glucose. The thoracic aorta was then dissected free of adherent connective tissue. The aortic segment was cut into rings of 3–4 mm in length, taking special care to avoid stretching or touching the luminal surface of the rings. The rings were suspended horizontally between two stirrups in organ chambers filled with 5 mL Krebs’ solution at 37 ºC and aerated continuously with 95% O2 and 5% CO2. One stirrup was connected to an anchor and the other was connected to a force transducer for recording isometric tension. The solution in the chambers was changed every 15-20 min. Rings were equilibrated for 60 min under 2 g resting tension and then challenged with 60 mmol/L KCl at least three times until a reproducible maximal contractile response was obtained. After repeated washing and a further equilibration period of 30 min, rings were contracted with a submaximal concentration of phenylephrine (1 µmol/L) and relaxed with cumulative concentrations of acetylcholine (0.03–3 µmol/L) at the plateau phase of the phenylephrine contraction to assess the integrity of the endothelium. The rings with the maximal relaxation (Emax) to 3 µmol/L acetylcholine of more than 80% were considered to be endothelium-intact and used in the study.
Experimental protocol After each ring was serially washed and re-equilibrated, the rings of the control group were incubated for 45 min with Krebs’ buffer. The rings of LPC group were incubated with Krebs’ buffer for 15 min and then exposed to LPC (1–10 µmol/L) for 30 min. In the pravastatin group, the rings were pre-incubated with various concentrations of pravastatin (0.3–3 mmol/L) for 15 min before exposure to 3 µmol/L LPC for 30 min, respectively. To determine whether increasing prostacyclin or NO production is involved in the protective effect of pravastatin on impaired endothelium-dependent relaxation induced by LPC, aortic rings were pretreated with indomethacin 10 µmol/L, an inhibitor of cyclooxygenase or L-NAME 30 µmol/L, an inhibitor of NOS in the presence of pravastatin (1 mmol/L) for 15 min and then exposed to 3 µmol/L LPC for 30 min.
To determine whether the activation of PKC, increased production of superoxide anions, and decreased NO synthesis contribute to the detrimental effects of LPC, rings were pre-incubated with chelerythrine (1 µmol/L), an inhibitor of PKC, SOD (200 kU/L), a scavenger of superoxide anions, or L-arginine (3 µmol/L), a precursor of NO for 15 min and then exposed to LPC (3 µmol/L) for another 30 min.
After the above incubations, all rings were re-contracted with 1 µmol/L phenylephrine and concentration responses to acetylcholine (0.03–3 µmol/L) were repeated. Before finishing the experiment, the relaxation response to sodium nitroprusside (10 µmol/L) at the plateau phase of the phenylephrine contraction was also tested in isolated aortic rings.
Statistical analysis Results are expressed as Mean± SEM. Relaxation and the maximal relaxation (Emax) produced by acetylcholine was calculated and expressed as the percentage of contraction elicited by phenylephrine. The half-maximal relaxation (EC50) response to acetylcholine was estimated by linear regression from log concentration-effect curves. Differences between groups were tested for statistical significance by analysis of variance followed by the Newman-Keuls test. P<0.05 was considered significant.
Results
Effects of LPC on endothelium-dependent relaxation There was no difference in the relaxation response to acetylcholine in rat aortic rings during the initial examination between groups (data not shown). After incubation with Krebs’ solution for 45 min, acetylcholine (0.03–3 µmol/L) still evoked a significant concentration-dependent relaxation in aortic rings in the control group during the later repeat examination (Figure 1), in which the Emax value reached 90.4%±4.3%, and the EC50 value was 74.9±9.6 nmol/L (Table 1). Exposure of aortic rings to LPC (1–10 µmol/L) for 30 min significantly inhibited the endothelium-dependent relaxation response to acetylcholine in a dose-dependent manner; there were lower Emax and higher EC50 values in aortic rings of the LPC groups compared with the control rings (Figure 1, Table 1). These results indicated that LPC impaired the endothelium-dependent relaxation response to acetylcholine in rat aortic rings.
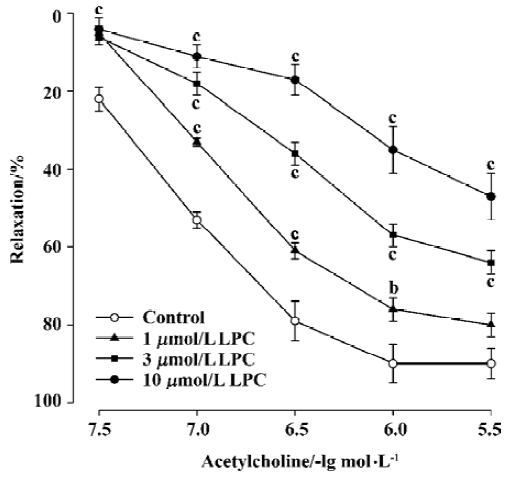
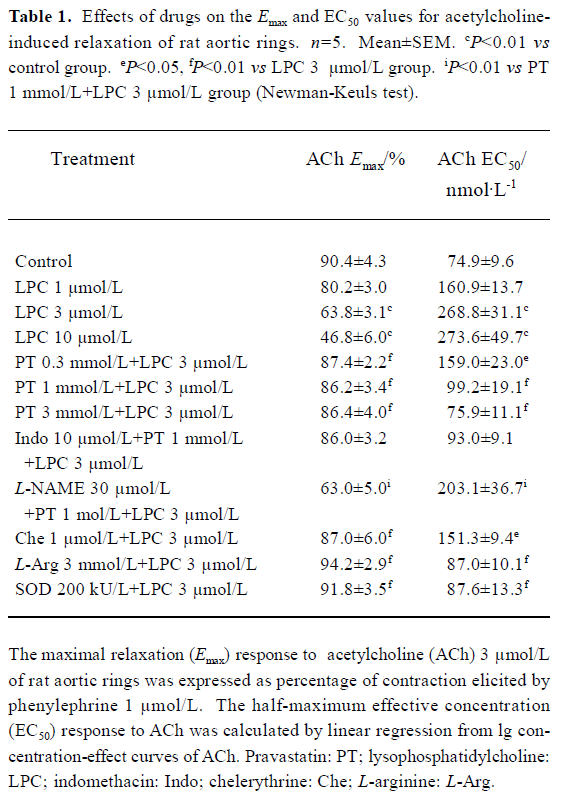
Full table
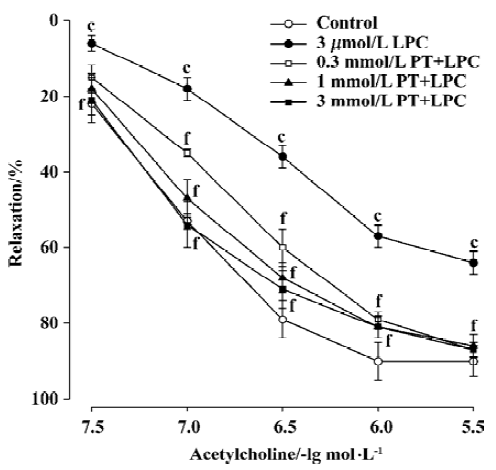
Effects of pravastatin on the inhibition by LPC Pre-incubation of rat aortic rings with pravastatin (0.3-3 mmol/L) for 15 min and then co-incubation of rat aortic rings with LPC 3 µmol/L for 30 min attenuated the inhibitory effect of LPC on endothelium-dependent relaxation. The Emax value increased and the EC50 value was reduced in aortic rings of pravastatin groups compared with the LPC 3 µmol/L group (Figure 2, Table 1).
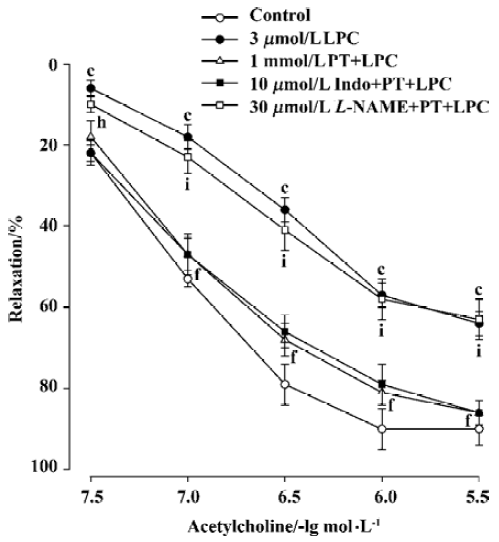
Incubation of aortic rings with L-NAME (30 µmol/L) in the presence of pravastatin (1 mmol/L) for 15 min abolished the protective effect of pravastatin on the impairment of endothelium-dependent relaxation induced by LPC, whereas indomethacin did not change the beneficial effects of pravastatin. Significant differences were obtained when the Emax and EC50 values of the L-NAME group, but not of the indomethacin group were compared with those of pravastatin group (Figure 3, Table 1). In addition, pravastatin per se did not affect the endothelium-dependent relaxation of aortic rings in the absence of LPC or endothelium-independent relaxation of aortic rings in the presence of LPC (data not shown).
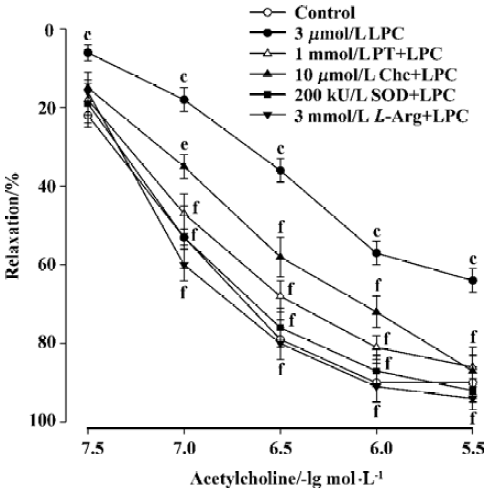
Effects of chelerythrine, L-arginine, and SOD on the inhibition by LPC In an analogous manner, pre-incubation of aortic rings with chelerythrine (1 µmol/L), L-arginine (3 mmol/L), or SOD (200 kU/L) also decreased the inhibition of endothelium-depedent relaxation by LPC. Significant differences were observed when the Emax and EC50 values of chelerythrine, L-arginine, or SOD group were compared with those of LPC group (Figure 4, Table 1).
Discussion
The present study confirmed previous reports that LPC inhibited endothelium-dependent relaxation of isolated rat aortas in a concentration-dependent manner[2], and demonstrated that pravastatin, an inhibitor of HMG-CoA reductase, reduced the inhibition by LPC on endothelium-dependent relaxation. The protective effect of pravastatin on the impairment of endothelium-dependent relaxation induced by LPC is very similar to that of chelerythrine, SOD, and L-arginine. These results suggest that pravastatin protects endothelium against injury from LPC and this may be related to enhancing the bioavailability of NO through increasing NO synthesis and decreasing NO oxidative inactivation in endothelial cells.
NO-mediated and endothelium-dependent relaxation is inhibited early in the atherosclerotic process, even before histological evidence of plaque formation is evident. Ox-LDL, an oxidative product of native LDL in endothelial cells and/or macrophages which presents in atherosclerotic lesions, is primarily responsible for this inhibition[1–3]. The inhibitory action of ox-LDL has generally been attributed to LPC, which is abundant in oxidized LDL and also accumulates in atherosclerotic lesions[1–3]. Removal of LPC from ox-LDL with phospholipase B[3] or albumin[19] has been shown to render ox-LDL inactive, and conversely, direct treatment of arterial rings with LPC can mimic the inhibitory effects of ox-LDL on endothelium-dependent relaxa-tion[2,3]. In the present study, we also confirmed that exposure of rat aortic rings to LPC (1–10 µmol/L) did inhibit the endothelium-dependent relaxation response to acetylcholine, but did not affect the endothelium-independent relaxation response to sodium nitroprusside. These results are consistent with other studies[2,3], and suggest that the concentrations of LPC used in the current study had endothelium-specific effects that were not due to its cytotoxicity.
Although the precise mechanisms responsible for the inhibition of endothelium-dependent relaxation induced by LPC have not been elucidated, increasing oxidative degradation of NO and decreasing production of NO in endothelial cells could be implicated in this inhibition. A number of studies[4,20] have suggested that LPC stimulates superoxide anion production by activating PKC in vascular cells, which can inactivate NO by oxidation. In addition, LPC has been shown to inhibit the high-affinity arginine transporter in endothelial cells, decrease the availability of arginine, and subsequently reduce the synthesis of NO; supplementation with exogenous L-arginine reversed the inhibition by LPC[21]. In the present study, we found that chelerythrine, an inhibitor of PKC, improved the impairment of endothelium-dependent relaxation by LPC. Furthermore, the current study also demonstrated that the inhibition of endothelium-dependent relaxation induced by LPC was completely reversed by SOD, a scavenger of superoxide anions and by L-arginine, a precursor of NO. Taken together, these findings strongly support the proposal that LPC-induced inhibition of endothelium-dependent relaxation may be due to decreasing the bioavailability of NO via increasing the oxidative degradation of NO and reducing the synthesis of NO.
Evidence has accumulated that ox-LDL plays a critical role in atherogenesis, and LPC, the major lipid constituent of ox-LDL, has been recognized as an important factor that triggers disturbed endothelium-dependent vasodilatation of atherosclerosis. Therefore, finding effective drugs to reverse endothelial dysfunction induced by LPC may prevent the development of atherosclerosis. Pravastatin, like the other statins, is a competitive inhibitor of HMG-CoA reductase, which markedly reduces hepatic cholesterol in vivo by inhibiting the rate-limiting step in cholesterol synthesis catalyzed by HMG-CoA reductase[5]. A number of studies have demonstrated that pravastatin can reduce adverse cardiovascular events in patients suffering from coronary artery disease with or without hypercholesterolemia[5,22]. Recently, some authors have reported that pravastatin improved the defective endothelium-dependent vasodilatation of atherosclerotic vessels before the reduction of plasma cholesterol levels in humans and animals[16–18]. However, it is still unknown that whether pravastatin acts directly against the adverse effect of LPC on endothelium-dependent relaxation in addition to its cholesterol-lowering action. This study provides the first evidence that pravastatin protects endothelium against injury induced by LPC in isolated rat aortas. Although experiments in vitro can not completely mimic the conditions in vivo and are not good for studying the chronic effects of drugs, they are useful because they can exclude the effects of many factors in vivo and directly investigate the effects of drugs in some (pathological) conditions. Therefore, the endothelium-dependent relaxation of isolated aortic rings is extensively used to measure the function of endothelium in physiological conditions and some pathological conditions, such as atherosclerosis, or mimic intervention with ox-LDL or LPC.
The mechanisms by which pravastatin improves endothelial function are not completely understood. Results from the present study indicate that the protective effect of pravastatin on vascular endothelium is related to increasing the synthesis of endothelial NO. We found that the protective effect of pravastatin on the inhibition of endothelium-dependent relaxation induced by LPC was similar to that of L-arginine. Furthermore, the beneficial effect of pravastatin was reversed by L-NAME, but not by indomethacin. Another study has reported that pravastatin can up-regulate the expression of NOS and enhance the production of NO in endothelial cells[23]. Treatment with statins including pravastatin of patients and rabbits with atherosclerosis results in up-regulated eNOS expression, increased NO synthesis, and simultaneously improved endothelium- dependent relaxation, which was reversed by Nω-nitro- L-arginine[24,25]. These findings suggested that pravastatin enhanced the production and release of NO from endothelium to improve endothelial function. However, Parker et al[26] reported that treatment of normal rats with simvastatin, but not with pravastatin, impaired the endothelium-dependent relaxation of their isolated aortic rings although it was able to enhance eNOS expression in endothelial cells. Chen et al[27] reported that pravastatin did not influence IL-1β-induced NO synthesis in vascular smooth muscle cells. The discrepancies between our findings and theirs could be attributed to the different functional states of vascular endothelium, in different cells and under treatment with different stimulated factors or different drugs. In fact, many studies have shown that pravastatin did increase NO production and restore endothelium-dependent relaxation in atherosclerotic animals and humans[16–18,25]. Another potential mechanism by which pravastatin improved endothelial function is by reducing the oxidative inactivation of NO. Recent investigations have demonstrated that statins can decrease lipid peroxidation production[7], suppress NADPH oxidase activity[28], and increase the activity of catalase[29], and superoxide dismutase[7], which decreases the generation of oxygen free radicals resulting in reduction of NO activation. Moreover, a number of studies have shown that pravastatin can also reduce superoxide anion production by suppressing PKC activity in vascular cells[14,15]. In the present study, we demonstrated that chelerythrine, an inhibitor of PKC and SOD, a scavenger of superoxide anions, reversed the inhibition of endothelium-dependent relaxation induced by LPC, which was similar to effects of pravastatin. These results indirectly suggest that the protective effect of pravastatin against the damage to endothelium induced by LPC is related to decreasing NO oxidative inactivation via suppressing PKC activity and reducing superoxide anion production.
In conclusion, the present study demonstrates for the first time that pravastatin has a protective effect on endothelium against injury elicited by LPC, and that this protective effect is related to enhancing the bioavailability of NO through increasing NO synthesis and decreasing NO oxidative inactivation in endothelial cells.
Footnote
Project supported by grants from the National Natural Science Foundation of China (N
References
- Yla-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest 1989;84:1086-95.
- Kugiyama K, Kerns SA, Morrisett JD, Roberts R, Henry PD. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature 1990;344:160-2.
- Mangin EL Jr, Kugiyama K, Nguy JH, Kerns SA, Henry PD. Effects of lysolipids and oxidatively modified low density lipoprotein on relaxation of rabbit aorta. Circ Res 1993;72:161-6.
- Ohara Y, Peterson TE, Zheng B, Kuo JF, Harrison DG. Lysophos-phatidylcholine increases vascular superoxide anion production via protein kinase C activation. Arterioscler Thromb 1994;14:1007-13.
- Koizumi J, Shimizu M, Miyamoto S, Origasa H, Mabuchi H. Effect of pravastatin-induced LDL-cholesterol reduction on coronary heart disease and cerebrovascular disease in Japanese: Hokuriku lipid coronary heart disease study-pravastatin atherosclerosis trial (Holicos-PAT). J Atheroscler Thromb 2002;9:251-9.
- Corti R, Fuster V, Fayad ZA, Worthley SG, Helft G, Smith D, et al. Lipid lowering by simvastatin induces regression of human atherosclerotic lesions: two years’ follow-up by high-resolution noninvasive magnetic resonance imaging. Circulation 2002;106:2884-7.
- Ma FX, Liu LY, Xiong XM. Protective effects of lovastatin on vascular endothelium injured by low density lipoprotein. Acta Pharmacol Sin 2003;24:1027-32.
- Jiang JL, Jiang DJ, Tang YH, Li NS, Deng HW, Li YJ. Effect of simvastatin on endothelium-dependent vasorelaxation and endogenous nitric oxide synthase inhibitor. Acta Pharmacol Sin 2004;25:893-901.
- Laufs U, Gertz K, Huang P, Nickenig G, Bohm M, Dirnagl U, et al. Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke 2000;31:2442-9.
- Sindermann JR, Fan L, Weigel KA, Troyer D, Muller JG, Schmidt A, et al. Differences in the effects of HMG-CoA reductase inhibitors on proliferation and viability of smooth muscle cells in culture. Atherosclerosis 2000;150:331-41.
- Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation 2002;105:739-45.
- Kishi T, Hirooka Y, Mukai Y, Shimokawa H, Takeshita A. Atorvastatin causes depressor and sympatho-inhibitory effects with upregulation of nitric oxide synthases in stroke-prone spontaneously hypertensive rats. J Hypertens 2003;21:379-86.
- Kagota S, Yamaguchi Y, Nakamura K, Kunitomo M. Functional evidence for anti-oxidant action of fluvastatin on low-density lipoprotein using isolated macrophages and aorta. Clin Exp Pharmacol Physiol 2000;27:401-5.
- Wagner AH, Kohler T, Ruckschloss U, Just L, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 2000;20:61-9.
- Yasunari K, Maeda K, Minami M, Yoshikawa J. HMG-CoA reductase inhibitors prevent migration of human coronary smooth muscle cells through suppression of increase in oxidative stress. Arterioscler Thromb Vasc Biol 2001;21:937-42.
- Wassman S, Faul A, Hennen B, Scheller B, Bohm M, Nickenig G. Rapid effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition on coronary endothelial function. Circ Res 2003;93:98-103.
- Silva EP, Fonseca FA, Ihara SS, Izar MC, Lopes IL, Pinto LE, et al. Early benefits of pravastatin to experimentally induced atherosclerosis. J Cardiovasc Pharmacol 2002;39:389-95.
- Kobayashi T, Matsumoto T, Kamata K. Mechanisms underlying the chronic pravastatin treatment-induced improvement in the impaired endothelium-dependent aortic relaxation seen in streptozotocin-induced diabetic rats. Br J Pharmacol 2000;131:231-8.
- Vuong TD, Kimpe SD, Roos RD, Rabelink TJ, Koomans HA, Joles JA. Albumin restores lysophosphatidylcholine-induced inhibition of vasodilation in rat aorta. Kidney Int 2001;60:1088-96.
- Murohara T, Kugiyama K, Ota Y, Doi H, Ogata N, Ohgushi M, et al. Effects of atrial and brain natriuretic peptides on lysophosphatidyl-choline-mediated endothelial dysfunction. J Cardiovasc Pharmacol 1999;34:870-8.
- Kikuta K, Sawamura T, Miwa S, Hashimoto N, Masaki T. High-affinity arginine transport of bovine aortic endothelial cells is impaired by lysophosphatidylcholine. Circ Res 1998;83:1088-96.
- Hague W, Forder P, Simes J, Hunt D, Tonkin A. LIPID Investigators. Effect of pravastatin on cardiovascular events and mortality in 1516 women with coronary heart disease: results from the Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) study. Am Heart J 2003;145:643-51.
- Kaesemeyer WH, Caldwell RB, Huang J, Caldwell RW. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J Am Coll Cardiol 1999;33:234-41.
- Thakur NK, Hayashi T, Sumi D, Kano H, Tsunekawa T, Iguchi A. HMG-CoA reductase inhibitor stabilizes rabbit atheroma by increasing basal NO and decreasing superoxide. Am J Physiol Heart Circ Physiol 2001;281:H75-83.
- Masumoto A, Hirooka Y, Hironaga K, Eshima K, Setoguchi S, Egashira K, et al. Effect of pravastatin on endothelial function in patients with coronary artery disease (cholesterol-independent effect of pravastatin). Am J Cardiol 2001;88:1291-4.
- Parker RA, Huang Q, Tesfamariam B. Influence of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors on endothelial nitric oxide synthase and the formation of oxidants in the vasculature. Atherosclerosis 2003;169:19-29.
- Chen H, Xing Y, Liu RH. Lovastatin increases nitric oxide synthesis in IL-1β- stimulated smooth muscle cells. Chin Med J 2001;114:1123-7.
- Bandoh T, Sato EF, Mitani H, Nakashima A, Hoshi K, Inoue M. Antioxidative potential of fluvastatin via the inhibition of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. Biol Pharm Bull 2003;26:818-22.
- Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 2002;22:300-5.