
Alteration of binding sites for [3H]P1075 and [3H]glibenclamide in reno-vascular hypertensive rat aorta1
Introduction
ATP-sensitive K+ channels(KATP) have been identified in multiple cells[1,2]. In vascular smooth muscles, KATP can regulate vascular tone[3]. The functions of vascular KATP can be modulated by nucleotides, potassium channel blockers, sulfonylureas, such as glibenclamide (Gli), and KATP openers, such as pinacidil (Pin)[4]. Recent molecular biological and electrophysiological studies on recombinant KATP have identified that KATP is a hetero-octameric complex composed of an inwardly rectifying K+ channel (Kir6.x) serving as a pore-forming subunit and regulatory sulfonylurea receptors (SURs) belonging to the ATP-binding cassette protein superfamily[5]. The binding sites for nucleotides, sulfonylureas, and potassium channel openers are localized in the SURs, which explains the complex sensitivity to these drugs[6]. Furthermore, it has been found that binding experiments have been carried out in vascular smooth muscles, using [3H]Gli and [3H]P1075, that the binding sites of KATP openers are different from those of blockers, and negatively allosteric interaction exists between these two binding sites[7,8]. The functional alterations of KATP in vascular smooth muscles from hypertensive animals have been investigated by using patch-clamp techniques; the dose-response curves of the KATP openers pinacidil and aprikalim, for inducing relaxation of isolated aorta and basilar arteries, were shifted rightward[9], and the effects of levcromakalim on KATP currents in mesenteric arteries were decreased in SHR[10]. But the mechanisms underlying the hypertensive changes of KATP remain to be investigated.
In the present study, the characteristics of [3H]P1075 and [3H]Gli binding in aortic strips, derived from normotensive and hypertensive rats, were compared for investigation of the hypertensive changes of KATP in vascular smooth muscles.
Materials and methods
Drugs [3H]Gli (specific activity 1850 GBq/mmol=50 Ci/mmol ) was purchased from Dupont/ New England Nuclear (Boston, MA, USA). [3H]P1075 (specific activity 121 Ci/mmol) from Amersham International (Buckinghamshire, United Kingdom). The radiolabels were stored in ethanol at -20 ºC. Gli, Pin, and Tris from Sigma Chemical Co (St Louis, MO, USA). HEPES from Boehringer Mannheim (Baden-Wurttemberg, Germany). Dimehylsulphoxide (Me2SO) from Merck Company (West point, USA). P1075 was kindly donated by Leo Pharmaceuticals (Ballerup. Denmark). All other chemicals and reagents were of analytical grade. Both P1075 and Pin were dissolved in Me2SO (25 g/L) and were diluted as appropriate so that the final concentration of Me2SO was less than 0.1%.
Animals Wistar rats (NWR, male, weighing 235±21 g, n=106), were provided by the Laboratory Animal Center of Institute of Pharmacology and Toxicology, Academy of Military Medical Sciences, Beijing, China.
Reno-vascular hypertension Reno-vascular hypertension was produced by coarctation of the abdominal aorta between the origin of the renal arteries, under ether anesthesia, as described by Liu et al[11]. The rats were used 30 d after the surgery. One day before the experiment, a polyethylene catheter was inserted into the carotid artery for blood pressure measurements with a two-channel physiological recorder (LMS-LB). Mean arterial pressure was 202±25 mmHg in RVHR and 140±11 mmHg in NWR. All animals were kept in a temperature-controlled room on a 14 h/10 h light/dark cycle and had free access to a standard rat chow diet and tap water.
Aorta strips All rats (NWR, n=55; RVHR, n=51) were decapitated and exsanguinated. Aorta strips were prepared as described in a previous study[12]. The preparations were then weighed and assigned into tissue holders and immersed in ice-cold HEPES-buffered physiological salt solution (PSS) containing (in mmol/L): NaCl 139, KCl 5, CaCl2 25, MgCl2 1.2, glucose 11, HEPES 5, gassed with 95% O2/5% CO2 and titrated to pH 7.40 with NaOH at 37 ºC.
Equilibrium binding experiments
Saturation assay Binding assays of [3H]P1075 or [3H]Gli were routinely performed in duplicate in 0.50 mL PSS containing 3–5 mg wet weight of aorta strips at 25 ºC or 37 ºC. Nonspecific binding, determined in the presence of 50 µmol/L unlabeled P1075 or 30 µmol/L Gli, accounted for 48.6%±6.4% or 44%±6% of total bound ligand under the conditions employed. Specific binding was obtained by subtracting the nonspecific binding from the total binding.
For saturation experiments, aorta strips were incubated with different concentrations of label (0.25-30 nmol/L) for 90 min. They were then washed by adding 9 mL of ice-cold quench solution (NaCl 154 mmol/L, Tris 50 mmol/L, pH 7.40) for 1 min, blotted, and dissolved hermetically over 2 h in 50 µL of 60% perchloric acid and 100 µL of 30% H2O2 at 80 ºC. The samples were supplemented with 2.5 mL of ethylene glycol ethyl ether and 5 mL of dimethylbenzene containing 1% 2-(4-tert-butylphenyl)-5-(4-biphenylyl)-1,3,4-oxadiazole for 8 h. Radioactivity was determined with liquid scintillation spectrometry (at an efficiency of 42%-55%). The equilibrium dissociation (KD) and the maximum number of binding sites (Bmax) of label were determined by Scatchard analysis, and Hill coefficient (nH) was calculated from Hill equation.
Displacement assay Aorta strips were incubated with 5 nmol/L [3H]P1075 or 3 nmol/L [3H]Gli and unlabeled inhibitors of interest, then washed and determined as described above. Specific binding of [3H]P1075 or [3H]Gli in the presence of unlabeled drugs was normalized to percentage of specific binding in the absence of these drugs. The concentration of drug inhibiting 50% of specific labeled-ligand binding (IC50) was analyzed using the weighted non-linear least-squares regression program. This program fits data assuming the presence of one or more binding sites and allows comparison of the relative goodness-of-fit using F-test. A two-site fit was accepted as superior to one-site fit only if significance exceeded P<0.05.
Kinetic assay To determine the association kinetics, the aorta strips were incubated with 3 nmol/L [3H]Gli or 5 nmol/L [3H]P1075 and aliquots were withdrawn at various time points for separation of bound and free ligand as described above. To measure the dissociation kinetics, the labels were equilibrated with aorta strips for 90 min. Dissociation was initiated by addition of a large excess of unlabeled ligands (Gli 30 μmol/L, P1075 50 μmol/L) and bound-label was determined at various time intervals ranging from 1 to 90 min. The constants of association and dissociation were calculated according to equations (1) LN[Beq·(B·B)-1]=Kapp·t, (2) Kapp=K1·[L]+K2, and (3) LN(B·Beq-1)=-K2·t where B represents the amount of specific [3H]Gli or [3H]P1075 binding at different time points, Beq denotes the amount of the receptor-label complex at equilibrium and Kapp the apparent rate constant of association, which depends on the rate constants of the association and dissociation (K1, K2), and on the concentration of labeled ligand employed ([L]).
Statistical analysis Results are expressed as mean±SD. Statistical significance of differences between groups of means was assessed by ANOVA followed by Dunnet’s test using the Instat Programme (Graphpad SAS Software, San Diego, USA) and was accepted at the P<0.05 level.
Results
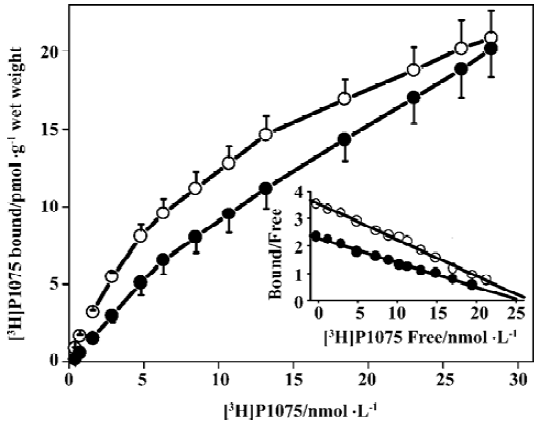
[3H]P1075 bindings [3H]P1075 bound in a saturated manner to aorta preparations in NWR and RVHR within the concentration range employed. Scatchard analysis of the data suggested that in NWR aorta strips [3H]P1075 bound to a single class of sites with a KD value of 7.7±2.0 nmol/L, and Bmax value of 26±8 pmol/g wet weight. In RVHR, [3H]P1075 also bound to a single class of sites. The KD value, however, increased ~1.5-fold and the Bmax value showed no change compared with those in NWR (Figure 1, Table 1).

Full table
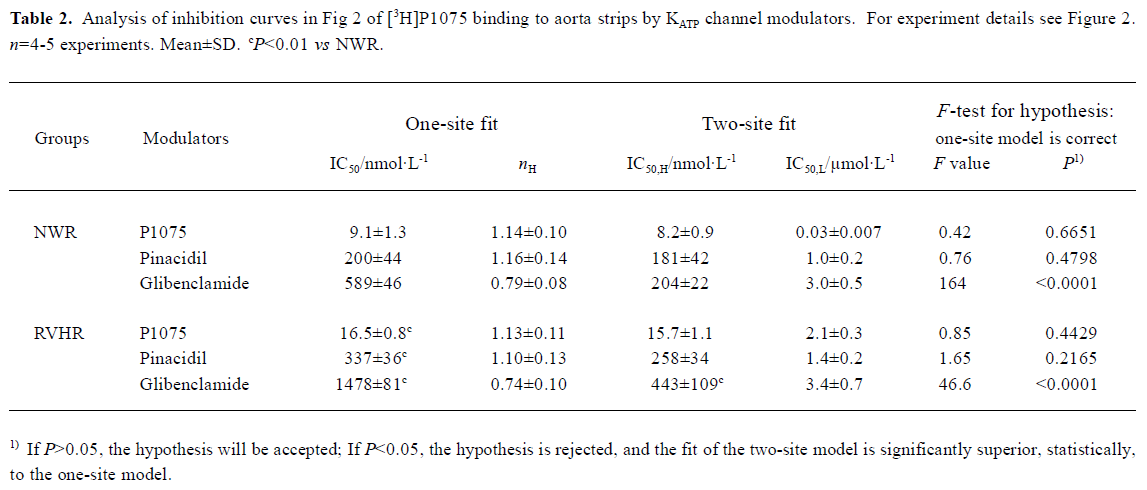
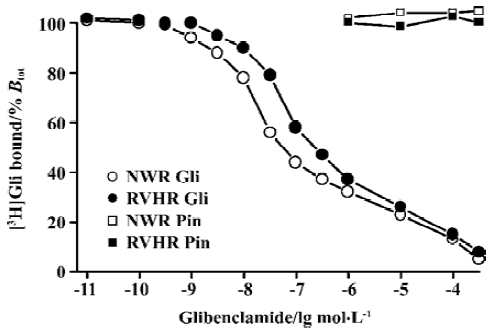
KATP opener P1075 and Pin, as well as blocker Gli, concentration-dependently inhibited specific [3H]P1075 binding. However, only KATP opener P1075 and Pin were able to inhibit the specific [3H]P1075 binding completely, whereas inhibition at saturation by Gli was incomplete only by up to 72% of maximal specific binding in NWR and RVHR aorta preparations. Compared with those in NWR, inhibition curves of [3H]P1075 binding to aorta strips in RVHR by these modulators were shifted to the right along the concentration axis (Figure 2). Statistical analysis with the F-test showed that the fit of inhibition curves by P1075 and Pin to the one-site model was significantly superior to two-site model, which corresponded to their nH being close to unity. IC50 for the inhibition of [3H]P1075 binding to RVHR aorta preparations by P1075 and Pin were increased by approximately 1.8- and 1.7-fold, respectively, compared with those in NWR. The change in IC50 value for inhibition of [3H]P1075 binding by P1075 in RVHR aorta strips was in fair agreement with that in the KD value obtained in the saturation equilibrium experiments in Figure 1.
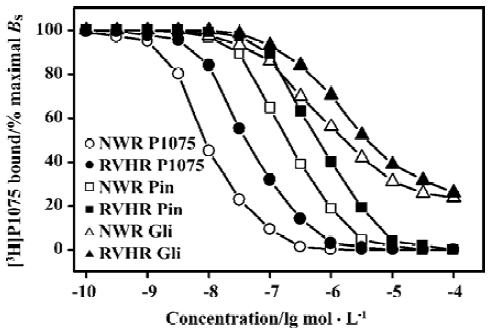
Inhibition of [3H]P1075 binding by Gli exhibited a biphasic plot (Figure 2) with nH significantly different from unity (Table 3). In addition, statistical analysis with the F-test showed the fit of inhibition curve by Gli to the two-site model was significantly superior to one-site model (Table 2). These results suggested that two binding sites for Gli were present. The IC50 values for the high- and low-affinity binding sites of Gli were shown in Table 2. Their respective nH values were found to be close to unity (not shown). IC50 value for inhibition of [3H]P1075 binding in RVHR aorta strips by Gli at high affinity site increased by more than 2-fold compared with that in NWR aorta preparation, but there was no significant difference between IC50 values at the low affinity site in RVHR and in NWR (Table 2).
Full table
Full table
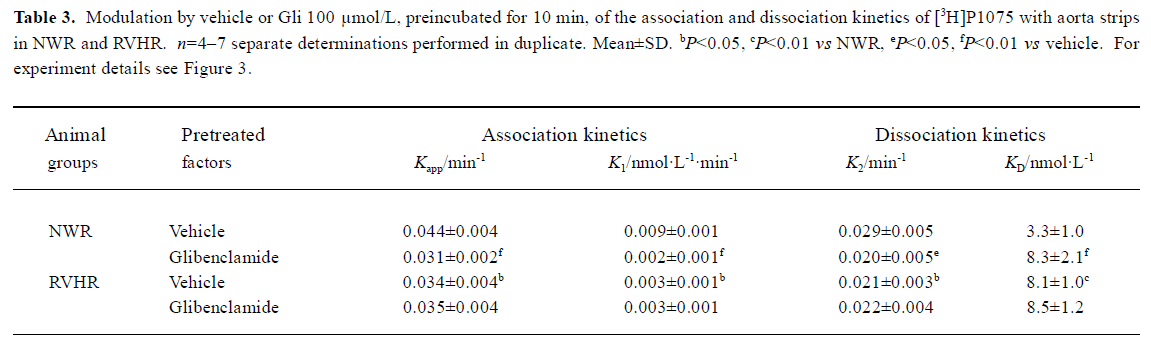
The kinetics of association and dissociation of [3H]P1075, measured at large excess of label over binding sites, are illustrated in Figure 3. In NWR, association kinetics of [3H]P1075 at 5 nmol/L followed a pseudo first-order kinetics, and the fit of this kinetics to equation 1 yielded a Kapp value of 0.044±0.004 min-1. After equilibrium was reached, dissociation of the receptor-label complex was initiated by addition of 50 μmol/L unlabeled P1075. The dissociation also followed a pseudo first-order kinetics with a K2 value of 0.029±0.005 min-1. Using equation 2, these values of Kapp and K2 allowed calculation of the second order rate constant of association, K1 to 0.009±0.001 nmol·L-1·min-1. From these rate constants, KD value of 3.3±1.0 nmol/L, where the large error in this value is explained by the propagation of errors, was calculated.
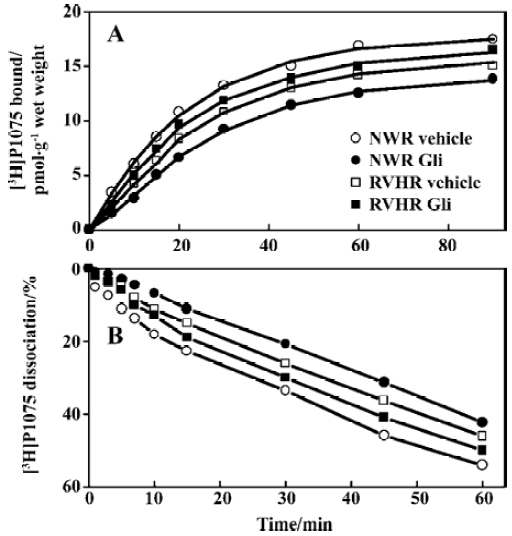
After NWR aorta strips were pretreated with Gli 100 μmol/L for 10 min, association and dissociation kinetics of [3H]P1075 still followed the pseudo first-order kinetics. However, association and dissociation were slowed down with the reduced Kapp and K2 value, but with increased KD value compared to those in vehicle (Table 3).
In RVHR, the association and dissociation of [3H]P1075 with aorta strips were also fitted the pseudo first-order kinetics, but slower association and dissociation kinetics were observed with increased KD value compared to those in NWR. Gli at 100 μmol/L preincubated with aorta strips for 10 min, did not alter the association and dissociation of [3H]P1075 with RVHR aorta strips (Figure 3, Table 3).
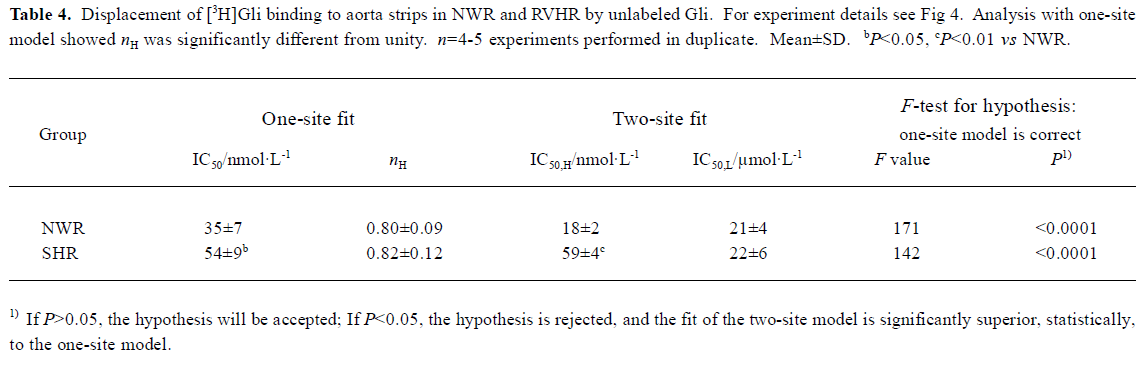
[3H]Gli binding experiments The competitive inhibition curve of 3 nmol/L [3H]Gli binding to aorta strips in NWR and RVHR by unlabeled Gli exhibited biphasic plots with nH significantly different from unity (Figure 4, Table 4). Inhibition curves were fitted with the one-site and two-site model, respectively. Statistical analysis with the F-test showed that the fit of curves of the two-site model was significantly superior to the one-site model (Table 4). Thus, two distinct binding sites for [3H]Gli were present in NWR and RVHR aorta preparations. In NWR aorta strips, the IC50 value for the inhibition of [3H]Gli binding at the high- and low-affinity sites by unlabeled Gli was 18±2 nmol/L and 21±4 μmol/L, respectively. The fit of one-site model gave nH equal to 0.80±0.09; a value in favor of heterogeneous class of binding sites with a negative cooperativity. In RVHR aorta strips, the IC50 value for inhibition of [3H]Gli binding at low-affinity sites by unlabeled Gli showed no difference from that in NWR aorta preparations, but IC50 at high-affinity sites increased approximately 4 fold. Pin, at concentrations up to 500 μmol/L neither inhibited the [3H]Gli binding to aorta strips in NWR nor in RVHR (Figure 4).
Full table
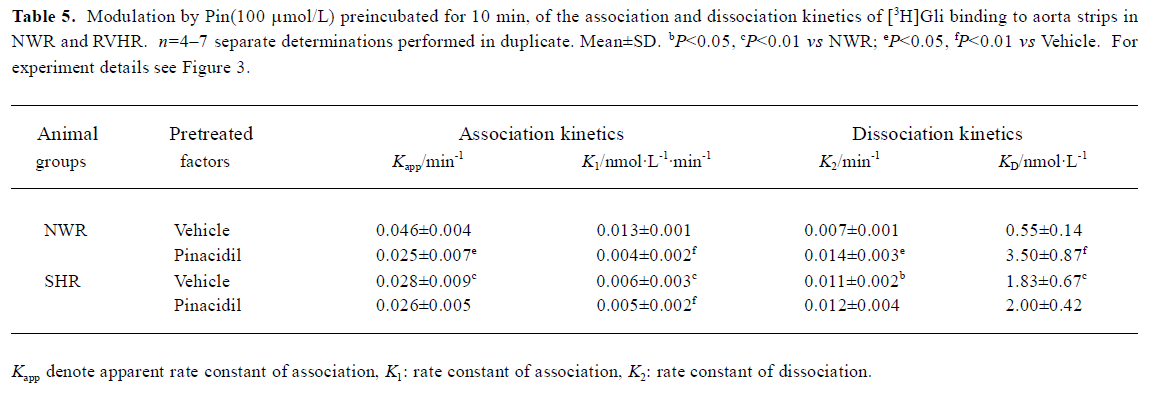
[3H]Gli binding to NWR aorta strips was rapid and approached equilibrium after a 40-50 min incubation period at 25 ºC (data not shown). After equilibrium was reached, the dissociation of [3H]Gli-receptor complex was initiated by the addition of 30 μmol/L unlabeled Gli. Association and dissociation of [3H]Gli followed pseudo first-order kinetics. The fit of the association and dissociation to equation 1 and 3 yielded Kapp and K2 of 0.046±0.004 and 0.007±0.001 min-1, respectively. After NWR aorta strips were pretreated with Pin (100 μmol/L) for 10 min, association and dissociation of [3H]Gli still followed pseudo first-order kinetics. However, when the association was retarded, Kapp was reduced by 45.6%, but when the dissociation was accelerated, K2 was increased approximately 2 fold (Table 5).
Full table
In RVHR aorta strips, association and dissociation of [3H]Gli were also fitted pseudo first-order kinetics, but when association became slower, Kapp was reduced by 39%, but when dissociation was faster, K2 was increased 1.5-fold. From these kinetics, the KD value calculated was increased 3.3-fold compared with those in NWR aorta strips. After RVHR aorta strips were pretreated with Pin 100 μmol/L for 10 min, the association and dissociation of [3H]Gli were not altered. From these kinetics, the KD value calculated was also unchanged (Table 5).
Discussion
Comparison of [3H]P1075 binding in NWR with that in RVHR In the present study, saturation experiments in NWR showed that aorta strips [3H]P1075 bound to a single class of sites with high affinity (KD=7.7±2.0 nmol/L) and relatively low density (Bmax=26±4 nmol·g-1 wet weight). These results are similar to those reported by Quast et al[13]. The KD value determined here corresponded well to the potency of P1075 as a vasorelaxant agent[14]. The KATP opener P1075 and Pin, as well as blocker Gli, inhibited [3H]P1075 binding according to the Law of Mass Action. The IC50 value (9.1±1.3 nmol/L) for inhibition of [3H]P1075 binding by P1075 was in fair agreement with the KD value obtained in the saturation experiments. The association and dissociation of [3H]P1075 with NWR aorta strips were time-dependent and monoexponential, these kinetic experiments allow a calculation of KD value of 3.3±1.0 nmol/L in reasonable agreement with the result of the saturation experiments (Table 1) and of competitive inhibition experiments (Table 2).
In the present study, [3H]P1075 binding in RVHR aorta strips was reported. The binding data showed that [3H]P1075 binding sites in RVHR aorta strips had similar qualities to those in NWR aorta preparations. However, some differences in [3H]P1075 binding in RVHR aorta strips were found: (1) KD value of [3H]P1075 binding increased approximately 1.5-fold; (2) IC50 value for the inhibition of [3H]P1075 binding by P1075 and Pin increased by 1.8- and 1.7-fold, respectively; (3) association and dissociation became slower. These results suggest that the affinity of [3H]P1075 binding sites to KATP openers is reduced in RVHR aorta strips. Therefore, we suggest that chronic hypertension might reduce the affinity of binding sites for KATP openers in aorta. Furthermore, the Bmax value of [3H]P1075 binding to aorta strips and the maximum inhibition of [3H]P1075 binding by P1075 and Pin showed no difference between RVHR and NWR. It is suggested that the number of [3H]P1075 binding sites is not altered in RVHR aorta strips. The present findings might explain why the dilatation of isolated aorta precontracted with norepinephrine or KCl in response to activation of KATP channels by Pin was impaired in SHR, why dilatation of the basilar artery by aprikalim was impaired in stroke-prone SHR in vivo[9]; and why the action of levcro-makalim on KATP channels in mesenteric artery muscle cells of SHR was decreased compared to normotensive rat cells[10]. The present findings also exclude the possibility that differences in response to Pin, aprikalim and levcromakalim are related to genetic differences between normotensive rats and SHR that are unrelated to hypertension.
Comparison of [3H]Gli binding in NWR with RVHR In the present study, the binding of [3H]Gli in NWR aorta strips was specific and reversible. Using a wide range of concentrations for Gli, we obtained evidence for the presence of both high and low affinity Gli-binding sites in aorta preparations: the former ranging from 1×10-11 to 1×10-7 mol/L with nanomolar affinity constant, and the latter from 1×10-7 to 1×10-3.5 mol/L with micromolar affinity constant. Our results are consistent with those reported previously in retarding vascular smooth muscle[12].
[3H]Gli binding in these preparations had similar qualities to those in NWR. However, [3H]Gli exhibited slower association and faster dissociation with an increased KD value in RVHR than in NWR (Table 5). The IC50 value for the inhibition of [3H]Gli binding by Gli at the high affinity component was increased, but was unchanged at the low affinity component. This indicates that the affinity of high affinity sulfonylurea sites is decreased in RVHR aorta strips.
Interaction of binding sites for KATP openers and blockers in NWR In the present study, [3H]P1075 bound to a single site in NWR aorta strips, whereas [3H]Gli bound to both high and low affinity sites in NWR aorta strips. [3H]P1075 binding can be inhibited by opener P1075 and Pin, but not completely by blocker Gli, whereas [3H]Gli binding could be inhibited by Gli, but not by Pin. Furthermore, [3H]P1075 and [3H]Gli binding present a similar association rate (0.044 and 0.046 nmol·L-1·min-1) but different dissociation rate(0.029 and 0.007 nmol·L-1·min-1, respectively). The most plausible explanation for these results is that KATP openers and blockers bind to different binding sites. In addition, Pin, preincubated with NWR aorta strips, slowed down the association of [3H]Gli and sped up the dissociation of [3H]Gli-receptor complex. It is suggested that the binding sites for KATP channel opener Pin negatively allosterically couple to the binding sites for KATP channel blocker Gli. The plausible mechanism is that the binding of Pin to its sites in SURs brings about configuration alteration in the Gli-sites, which hinders the binding of [3H]Gli to its sites and facilitates faster release of the radiolabel from receptor-label complex. Likewise, Gli, preincubated with NWR aorta strips, also slowed down the association of [3H]P1075 and sped up the dissociation of [3H]P1075-receptor complex. It was indicated that the sites for KATP channel blocker Gli also negatively allosterically coupled to the sites for KATP channel opener P1075. Alternatively, Gli down-regulates the affinity of the P1075 binding sites through inducing its configuration alteration. The evidence for a negative allosteric coupling between the sites for P1075 and Gli has been previously obtained for the rat aorta[8]. In the experiments, however, rat aorta preparations were only labeled with [3H]P1075, and the dissociation of [3H]P1075-receptor complex was initiated by addition of an excess of Gli rather than P1075 after [3H]P1075 binding reached equilibrium. In the present study, aorta strips were pretreated with Gli, and dissociation was initiated by addition of an excess of P1075 rather than Gli. A negative allosterism between the sites for openers and blockers was identified in a different experimental protocol in this study.
Interaction of binding sites for KATP openers and blockers in RVHR In the present paper, we studied the interaction between the binding sites for KATP openers and blockers in RVHR aorta strips. The evidence that KATP openers and blockers bound to different binding sites was also obtained in RVHR. In contrast to NWR, pretreatment with Gli failed to alter the association and dissociation kinetics of [3H]P1075 with aorta strips in RVHR. It is suggested that the negative allosteric action of sulfonylurea Gli on the binding sites for the opener P1075 was damaged in RVHR aorta strips. Similarly, pretreatment of Pin did not alter the association and dissociation kinetics of [3H]Gli with RVHR aorta strips. It is indicated that the negative allosteric action of opener Pin on the binding sites for sulfonylurea Gli is also damaged in SHR aorta strips.
In conclusion, the affinity of P1075 binding sites and high-affinity sulfonylurea binding sites are reduced, and the negative allosteric interactions between the two binding sites are impaired in RVHR aorta strips.
Footnote
Project supported by the Key State 863 Project (N
References
- Ashcroft SJ, Ashcroft FM. Properties and functions of ATP-sensitive K-channels. Cell Signal 1990;2:197-214.
- Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol 1991;261:H1675-86.
- Jackson WF. Arteriolar tone is determined by activity of ATP-sensitive potassium channels. Am J Physiol 1993;265:H1797-803.
- Davies NW, Standen NB, Stanfield PR. ATP-dependent K+ channels of muscle cells: their properties, regulation, and possible functions. J Bioenerg Biomembr 1991;23:509-35.
- Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 1995;270:1166-70.
- Inagaki N, Gonoi T, Clement JP 4th, Wang CZ, Aguilar BL, Bryan J, et al. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive potassium channels. Neuron 1996;16:1011-7.
- Löffler C, Quast U. Pharmacological characterization of the sulphonylurea receptor in rat isolated aorta. Br J Pharmacol 1997;120:476-80.
- Bray KM, Quast U. Specific binding site for K+ channel openers in rat aorta. J Biol Chem 1992;267:11689-92.
- Kitazono T, Heistad DD, Faraci FM. ATP-sensitive potassium channels in the basilar artery during chronic hypertension. Hypertension 1993;22:677-81.
- Ohya Y, Setoguchi M, Fujii K, Nagao T, Abe I, Fujishima M. Impaired action of levcromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension 1996;27:1234-9.
- Liu J, He ZY, Xu SM, Liu FY, Wang PY. Inositol 1,4,5-triphosphate receptor (IP3Rs) in myocardial nuclei involved in pressure overload-induced hypertrophy of rat heart. Acta Physiol Sin 2001;53:281-5.
- Zhu QL, He HM, Xiao WB, Wang H. Modulation by nucleotides of binding sites for [3H]glibenclamide in rat aorta and cardiac ventricular membranes. J Cardiovasc Pharmacol 2001;37:522-31.
- Quast U, Bray KM, Andres H, Manley PW, Baumlin Y, Dosogne J. Binding of the K+ channel opener [3H]P1075 in rat isolated aorta: relationship to functional effects of openers and blockers. Mol Pharmacol 1993;43:474-81.
- Higdon NR, Khan SA, Buchanan LV, Meisheri KD. Tissue and species variation in the vascular receptor binding of 3H-P1075, a potent KATP opener vasodilator. J Pharmacol Exp Ther 1997;280:255-60.