
Neuropeptide Y expression in mouse hippocampus and its role in neuronal excitotoxicity1
Introduction
Excitotoxicity is a process by which glutamate or other excitatory amino acids induce neuronal cell death in the central nervous system[1,2]. It is thought that excitotoxicity might contribute to the pathogenesis of human neuronal cell loss by sustained epilepsy, trauma, ischemia, and hypoglycemia, and might also underlie certain neurodegenerative diseases[3–5]. Excessive stimulation of the three types of ionotropic glutamate receptors, including the N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and KA receptors, produces neuronal cell death by increasing intracellular Ca2+ and Na+, and therefore drastically changing normal cellular physiology[1,2].
KA is an analog for glutamate and is also an excitotoxin in the hippocampus[6–8] that induces ongoing convulsions by directly stimulating KA receptors and indirectly increasing the release of excitatory amino acids from nerve terminals, degeneration of cornu ammonis (CA) pyramidal neurons in the hippocampus, and hyper-excitability of surviving CA neurons[9–11]. Thus, KA-induced seizures have been widely used as an epilepsy model for studying mechanisms underlying both seizure susceptibility and neuronal cell loss[12–14].
Increasing evidence suggests that NPY is a powerful endogenous anticonvulsant in the hippocampus[12,15–17]. NPY knockout mice exhibit uncontrollable seizures and enhanced fatality in response to KA administration[12]. Moreover, intracerebroventricular infusion of NPY reduces epileptiform after discharges and inhibits KA-induced seizures[18]. The possible mechanism for the anticonvulsant properties of NPY is the inhibition of calcium influx and glutamate release[12,15,17]. However, the exact role of NPY in vivo in excitotoxicity is unknown. Although excitotoxicity induced an ongoing neuronal apoptosis, it was mainly determined at early stages after excitatory amino acids treatment[7,8,14]. Thus, to investigate the sequential and spatial expression of NPY in the hippocampus within early stage of KA injection, and to understand its role in excitotoxicity, could be of unique importance.
Materials and methods
Animals and drugs Mice of 8 to 9 weeks age were used for analysis. All mice were housed with food and water ad libitum in a 12-h light cycle room. Our protocol was approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University, and we strictly followed NIH guidelines for the care and use of laboratory animals. All efforts were taken to minimize both animal suffering and the number of mice used. KA (Sigma Chemical, St Louis, MO, USA) was dissolved in ddH2O, neutralized by NaOH and injected (ip) in a dose of 20 mg/kg of body weight to the mice. Human NPY, NPY13-36 (Y2 agonist), or human pancreatic polypeptide (hPP) (Y5 agonist) were purchased from Bachem (Torrance, CA, USA).
IHC analysis of NPY expression At 0 (normal control treated with saline), 2, 4, 8, 16, or 24 h (8 each group) after KA injection, surviving mice were deeply anesthetized with sodium pentobarbital (60 mg/kg of body weight, ip), and then transcardially perfused with phosphate buffered saline (PBS), pH 7.4, and 4% paraformaldehyde in PBS for 15 min. After perfusion, their brains were removed, postfixed overnight with 4% paraformaldehyde in PBS at 4 ºC, submerged in 20% sucrose at 4 ºC for 48 h, and then sections were cut coronally at 40 µm to encompass the entire hippocampus using a freezing microtome. Every eighth of serial coronary brain sections through the rostral-caudal extent of the hippocampus in 40-µm thickness were selected for immunohistochemistry (IHC) detection of NPY expression in the hippocampus using rabbit anti-NPY antibody (1:1,000, Chemicon, CA, USA) as previously described[14,19].
Probe preparation for in situ hybridization (ISH) analysis The plasmid construct pT7T3D-Pac, containing 561 bp mouse NPY gene, was from the University of Cincinnati Microarray Center. The construct was first linearized by EcoR I or Hind III respectively. The EcoR I linearized template containing T3 promoter was used for anti-sense probe transcription. The Hind III linearized template containing T7 promoter was used for sense probe transcription (negative control). Transcription reaction mixture 20 µL in a RNase free eppendorf tube [4 µL 5×transcription buffer (Promega, USA), 1 µL linearized DNA (1 µg), 2 µL NTP mix (Dig-UTP) (Roche, Germany), 2 µL 0.1 mol/L DTT, 9 µL H2O, 1 µL RNase inhibitor (Invitrogen, USA), 1 µL RNA polymerase (T3 or T7) (Promega, USA)] was incubated at 37 ºC for 2 h, then 2U DNase I (RNase free) (Promega) was added, incubated at 37 ºC for 15 min to digest DNA template. Reaction was stopped with 2 µL of 0.2 mol/L EDTA, pH 8.0. Then 2.5 µL LiCl 4 mol/L and 75 µL prechilled (-20 ºC) 100% EtOH was added and mixed well. It was placed at -20 ºC for 2 h and centrifuged at 2–8 ºC, 44 500×g, for 15 min. The supernatant was discarded carefully. Samples were dried under a vacuum and then dissolved for 30 min at 37 ºC in 100 µL diethyl-pyrocarbonate (DEPC) H2O. RNA probe was quantified by spectrophotometric analysis (OD260/OD280) and stored at -80 ºC, or stored in aliquots at -20 ºC.
ISH detection of NPY expression For ISH analysis, 0 (normal control), 4, 8, or 16 h (8 each group) after KA injection, mice were killed by cervical dislocation, brains were removed quickly and kept in dry ice powder for 5 min and then stored at -80 ºC for at least 48 h before sectioning. Brain sections of 12 µm, containing hippocampal formation, were cut and placed on Superfrost Plus slides (pretreated with 0.1% DEPC H2O for 2 h at 37 ºC) and air dried at room temperature for 30 min and then stored at -80 ºC before use. Every fifteenth of serial coronary brain sections through the rostral-caudal extent of the hippocampus were analyzed by ISH as described previously[20].
Cannula implantation and infusion of NPY and its agonists Mice were anesthetized with an injection of Equithesin (34 mg/kg of body weight, ip) and atropine (0.35 mg/kg of bodyweight, ip). Animals were placed in a stereotactic device (Kopf Instruments), and cannulas were implanted into the lateral ventricle (0.6 mm posterior; 1.9 mm lateral; and 2.0 mm ventral to bregma). Mice were allowed to recover from surgery for 2 d. First, to determine the timing of NPY infusion after KA injection, icv injection of human NPY (5.0 µg in 1 µL of neutral buffered solution) or saline was performed 2, 8, or 24 h after KA injection (8 to 10 each group) using an infusion pump (KD Scientific) at a constant rate of 1 µL/min as described[12]. Then, to determine the dose of NPY infusion, 8 h after KA injection, the effect of icv infusion of NPY at a dose of 2.5, 5.0, and 10.0 µg was compared. Finally, to investigate the receptors that mediate its role in excitotoxicity, icv infusion of 3.0 µg NPY13-36 or 5.0 µg hPP was carried out 8 h after KA injection. The dose of NPY and its agonists were based on previous studies[12,16]. Cannula placement was verified post-hoc in all animals by injection of 1% cresyl violet solution before brain dissection. Three days after KA injection, brain sections were made as IHC assay above for TUNEL staining.
TUNEL assays Every eighth of serial coronary brain sections through the rostral-caudal extent of the hippocampus in 40-µm thickness were used for TUNEL staining using the in situ Cell Death Detection Kit (TUNEL POD) (Roche Applied Science, Germany) as described[14].
Statistical analysis IHC and ISH signal is in every of sub-region of each section was obtained with densitometric scanning, and data were analyzed by one–way ANOVA. All TUNEL positive cells in every sub-region of each section were counted regardless of sizes and shapes. The mean number of cells per section was used for comparison. Data were analyzed by one-way ANOVA and post-hoc Student-Newman-Keuls (SNK) test. Statistical significance was set as P<0.05.
Results
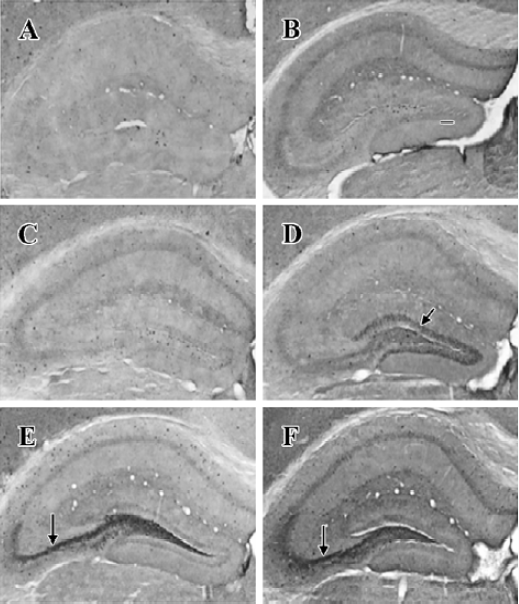
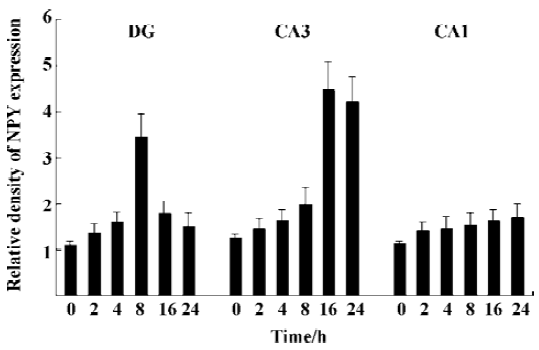
NPY expression in the hippocampus by IHC method In the granule cell layer of DG, no NPY positive cell was detected at basal condition (0 h) and 2 h after KA injection (Figure 1A, 1B). NPY positive cells appeared at 4 h (Figure 1C), reached the peak at 8 h (Figure 1D), and were reduced remarkably at 16 and 24 h (Figure 1E, 1F). In the CA3 sub-region, no positive signal was found within the first 4 h after KA injection (Figure 1A,1C), NPY positive signals appeared at 8 h (Figure 1D), strong signals appeared at 16 and 24 h (Figure 1E, 1F). No noticeable signal was detected in the CA1 sub-region at all time points within the first 24 h after KA injection. In the hilus, NPY positive signals were noticeably intensified at 8 h (Figure 1D), and reached the peak at 16 and 24 h (Figure 1E, 1F). One-way ANOVA results showed a significant time-dependent effect (P<0.05) in both DG and CA3 regions (Figure 2).
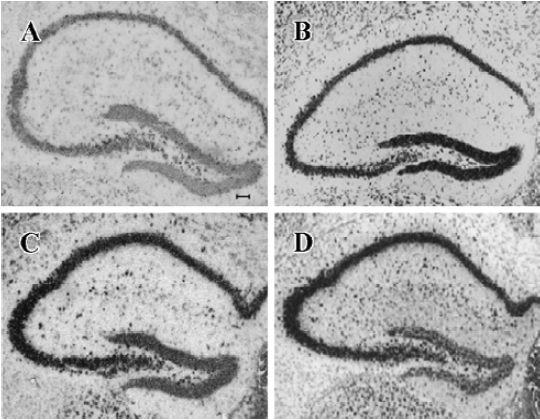
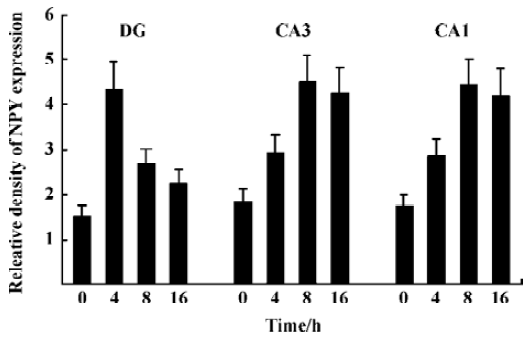
NPY expression in the hippocampus by ISH analysis Under physiological condition, NPY expression in the granule cell layer of DG, CA3, and CA1 was less noticeable (Figure 3A). Positive ISH signals were detected in DG, CA3, and CA1 sub-regions at each time point after KA treatment (Figure 3B–3D). Four hours after KA injection, ISH signals in the DG sub-region were stronger than those in CA3 and CA1 (Figure 3B). Eight and sixteen hours after KA injection, ISH signals were significantly reduced in DG sub-region but remarkably elevated in CA3, CA1, and hilus sub-regions (Figure 3C, 3D, P<0.05). One-way ANOVA result showed a significant time-dependent effect (P<0.05) in all regions (Figure 4).
NPY prevents neuronal cells from apoptosis in a time- and dose-related manner In CA3 and CA1, less massive apoptotic pyramidal cells were detected in mice with NPY infusion at 2 and 8 h after KA treatment (Figure 5A-5D) compared with control mice (Figure 5G, H). Similar TUNEL signals were detected in CA3 and CA1 between mice infused with NPY at 24 h after KA injection and control mice (Figure 5E-H). One-way ANOVA showed significant time-dependent effects in both CA3 and CA1 (P<0.05, Figure 6A). Post hoc SNK test showed a more noticeable effect at 2 h than that at 8 h of NPY infusion (P<0.05) though they both significantly reduced apoptosis in comparison with control mice (P<0.05) (Figure 6A). Figure 6B shows the quantitative results of TUNEL staining in CA3 and CA1 sub-regions 3 d after KA treatment with icv infusions of 2.5, 5.0, or 10.0 µg human NPY or saline (8 to 10 mice each group) at 8 h after KA injection. One-way ANOVA showed significant dose-dependent effect in both CA3 and CA1 among four groups (P<0.05). Post hoc SNK test showed a significant effect with infusion of 10.0 and 5.0 µg NPY at 8 h in comparison with control (P<0.05).
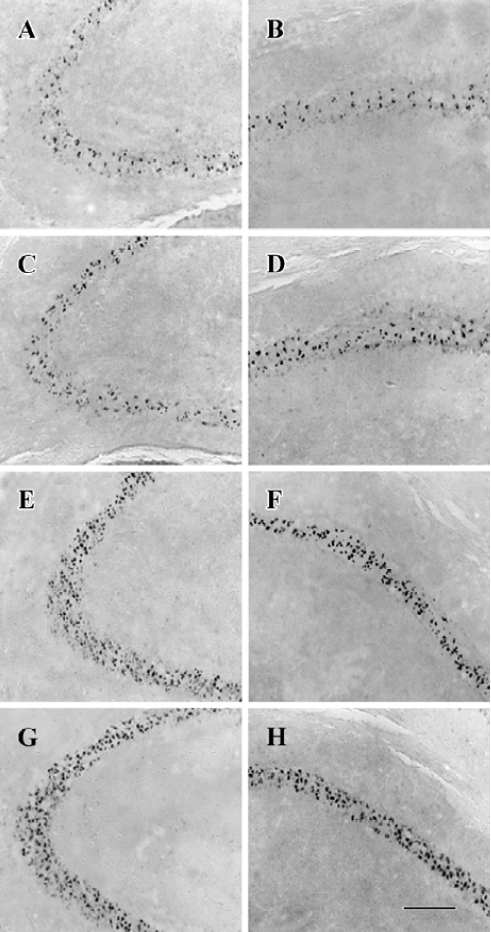
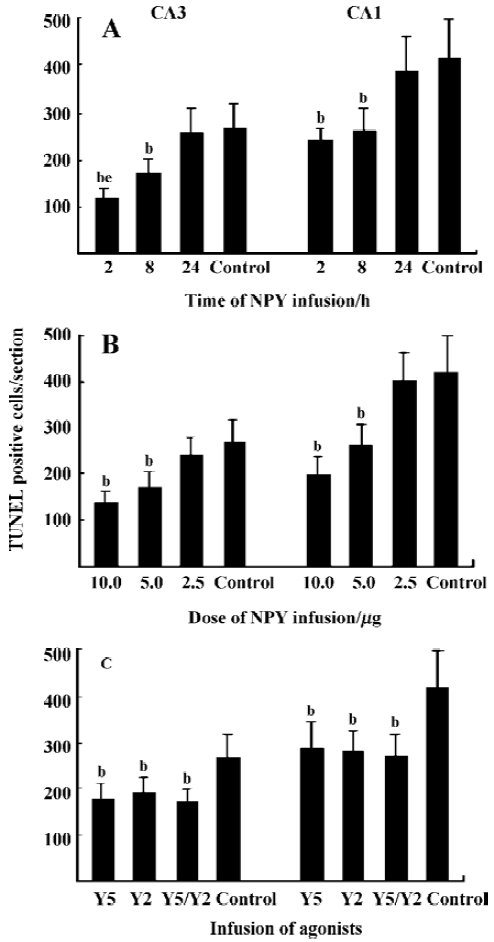
NPY prevents neuronal cells from apoptosis by activating both Y5 and Y2 receptors Y5 and Y2 agonists icv could both prevent neuronal cells from death 8 h after KA injection, but there was no additive effect between them. One-way ANOVA showed significant effects in both CA3 and CA1 (P<0.05). Post hoc SNK test showed no significant difference among Y5, Y2, and Y5/Y2 agonists treated three groups, but significant difference was detected between these three groups and control mice, respectively (Figure 6C, P<0.05).
Discussion
Increasing evidence suggests that NPY is a powerful endogenous anticonvulsant in the hippocampus[12,15–17]. Its expression can be elevated by experimentally induced seizures initiated by electrical kindling, kainic acid, and pentylenetetrazol[21-25]. Excitotoxicity induced an ongoing neuronal apoptosis, which was mainly determined at early stage after excitatory amino acid treatment[7,8,14]. According to these findings, we for the first time focused on NPY expression pattern in the hippocampus within early stages after KA injection using both IHC and ISH methods, and tried to understand its implication in excitotoxicity. From our IHC and ISH results (Figure 1–4), we concluded that NPY expression in the granule cell layer of DG sub-region was prior to CA3 and CA1 after KA-treatment, seizure-induced increase of NPY in the hippocampus was the result of transcriptional activation, and might be a compensatory mechanism to reduce excitatory neurotransmission. ICV infusion of NPY and its Y5, Y2 receptor agonists at 2 and 8 h after KA insult could rescue pyramidal neurons in CA3 and CA1 regions from apoptosis (Figure 3-6). It is also well established that the pyramidal cells in CA3 and CA1 are more susceptible to KA-induced apoptosis than the granule cells in DG[7,8,14]. Taken together, we presume that the preceded elevation of NPY expression in the granule cell layer of DG after KA injection might partially explain its different excitotoxicity-induced apoptotic responses in comparison with the pyramidal neurons from CA3 and CA1 regions.
Activation of NPY receptors is neuroprotective against excitotoxicity in rat hippocampal slice cultures[26]. For dentate granule cells and CA3 pyramidal cells, selective activation of Y1, Y2, or Y5 receptors, respectively had a neuro-protective effect against AMPA and KA, whereas only the activation of Y2 receptors was effective for CA1 pyramidal cells in the rat. Some physiological and pharmacological work showed intracerebroventricular administration of NPY reduced epileptiform afterdischarges and inhibited KA-induced seizures in rats; and Y5 and/or Y2 receptors were responsible for modulation of seizure activity in mouse hippocampal slices[18,27]. But, to date, whether NPY can prevent neuronal cells from excitotoxicity-induced apoptosis in vivo, what receptors are responsible for this function, what is the effective time and dose of NPY and its agonists infusion, are not known. We found icv infusion of NPY and its Y5, Y2 agonists within 8 h of a proper dose, after excitatory amino acid injection, could rescue neuronal cells from apoptosis in mice (Figure 5, 6). These data are quite useful for the potential clinical application of NPY in anti-epilepsy. The protective signal transduction pathway of its different receptors should be included in our future research.
With IHC detection, NPY positive or negative signal does not mean that NPY is synthesized or is not synthesized. As a neurotransmitter[28], NPY can be released from one cell where it is synthesized and be taken up by other cells through synaptic formation. Comparing the IHC result (Figure 1) with the ISH result (Figure 3), we found although NPY mRNA transcripts were synthesized in all regions of the hippocampus at 4, 8, and 16 h after KA treatment, no noticeable signals were detected in the CA1 region by IHC assay at any point. Probably, NPY in this region was transmitted to other places as a neurotransmitter after translation. Or the sensitivity of IHC is not high enough to detect NPY expression in this region.
In conclusion, the preceding elevated expression of NPY in granule cell layer of DG after KA injection may explain partially its different excitotoxicity-induced apoptotic responses in comparison with the pyramidal neurons from CA3 and CA1 regions. NPY can not only reduce neuronal excitability but also prevent excitotoxicity-induced neuronal apoptosis by activation of Y5 and Y2 receptors.
Footnote
Project supported by a grant from the National Natural Science Foundation of China (N
References
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988;1:623-34.
- Choi DW. Excitotoxic cell death. J Neurobiol 1992;23:1261-76.
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature 1999;399:A7-14.
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993;262:689-95.
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature 1999;399:A14-22.
- Campocdhiaro P, Coyle J. Ontogenetic development of kainite neurotoxicity: correlates with glutamatergic innervation. Proc Natl Acad Sci USA 1978;75:2025-9.
- Cook T, Crutcher K. Intrahippocampal injection of kainic acid produces significant pyramidal cell loss in neonatal rats. Neuroscience 1986;18:79-92.
- Nadler J, Perry B, Cotman C. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature 1978;271:676-7.
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985;14:375-403.
- Dudek F, Obenaus A, Schweitzer J, Wuarin J. Functional significance of hippocampal plasticity in epileptic brain: electrophysiological changes of the dentate granule cells associated with mossy fiber sprouting. Hippocampus 1994;4:259-65.
- Nadler J. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 1981;29:2031-42.
- Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci 1997;17:8927-36.
- Zhao WJ, Ma YH, Fei J, Mei ZT, Guo LH. Increase in drug-induced seizure susceptibility of transgenic mice overexpressing GABA transporter-1. Acta Pharmacol Sin 2003;24:991-5.
- Dong H, Csernansky CA, Goico B, Csernansky JG. Hippocampal neruogenesis follows kainic acid-induced apoptosis in neonatal rats. J Neurosci 2003;23:1742-9.
- Colmers WF, Bleakman D. Effects of neuropeptide Y on the electrical properties of neurons. Trend Neurosci 1994;17:373-9.
- Marsh DJ, Baraban SC, Hollopeter G, Palmiter RD. Role of the Y5 neuropeptide Y receptor in limbic seizures. Proc Natl Acad Sci USA 1999;96:13518-23.
- Vezzani A, Sperk G, Colmers W., Neuropeptide Y. Emerging evidence for a functional role in seizure modulation. Trends Neurosci 1999;22:25-33.
- Woldbye D, Madsen TM, Larsen PJ, Mikkelsen JD, Bolwig TG. Neuropeptide Y inhibits hippocampal seizures and wet dog shakes. Brain Res 1996;737:162-8.
- Wu Y, Zhang D, Lou D, Fan Y, Aronow D, Xu M, et al. c-fos regulates neuropeptide Y expression in mouse dentate gyrus. Neurosci Lett 2004;363:6-10.
- Braissant O, Wahli W. A simplified in situ hybridization protocol using non-radioactively labeled probes to detect abundant and rare mRNAs on tissue sections. Biochemica 1998;1:10-6.
- Marksteiner J, Sperk G, Maas D. Differential increases in brain levels of neuropeptide Y and vasoactive intestinal polypeptide after kainic acid-induced seizures in the rat. Naunyn Schmiedebergs Arch Pharmacol 1989;339:173-7.
- Pitkänen A, Beal MF, Sirviö J, Swartz KJ, Männistö PT, Riekkinen PJ. Somatostatin, neuropeptide Y, GABA and cholinergic enzymes in brain of pentylenetetrazol-kindled rats. Neuropeptides 1989;14:197-207.
- Tønder N, Kragh J, Finsen BR, Bolwig TG, Zimmer J. Kindling induces transient changes in neuronal expression of somatostatin, neuropeptide Y, and calbindin in adult rat hippocampus and fascia dentata. Epilepsia 1994;35:1299-308.
- Gruber B, Greber S, Rupp E, Sperk G. Differential NPY mRNA expression in granule cells and interneurons of the rat dentate gyrus after kainic acid injection. Hippocampus 1994;4:474-82.
- Kragh J, Tønder N, Finsen BR, Zimmer J, Bolwig TG. Repeated electroconvulsive shocks cause transient changes in rat hippocampal somatostatin and neuropeptide Y immunoreactivity and mRNA in situ hybridization signals. Exp Brain Res 1994;98:305-13.
- Silva AP, Pinheiro PS, Carvalho AP, Carvalho CM, Jakobsen B, Zimmer J, et al. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J 2003;10:122-31.
- Guo H, Castro PA, Palmiter RD, Baraban SC. Y5 receptors mediate neuropeptide Y actions at excitatory synapses in area CA3 of the mouse hippocampus. J Neurophysiol 2002;87:558-66.
- Wahlestedt C, Reis DJ. Neuropeptide Y-related peptides and their receptors-are the receptors potential therapeutic drug targets? Annu Rev Pharmacol Toxicol 1993;32:309-52.